Polymorphism of variable number tandem repeat gene of Mycobacterium tuberculosis isolates from Inner Mongolia
SU Yun-kai1, YU Qin2,3, LV Bing2, MA Yan3, LIAN Lu-lu2,3, YANG Xiao-min1, DONG Hai-yan2, LIU Yao1, ZHAO Xiu-qin2, WU Yi-mou3, WAN Kang-lin2,3
Inner Mongolia Institute for Tuberculosis Control and Prevention, huhehaote 010010, Inner Mongolia, China
Abstract
Objective To understand the polymorphism and proportion of variable number tandem repeat(VNTR) gene of Mycobacterium tuberculosis isolates from Inner Mongolia and the application of different VNTR loci for M. tuberculosis genotyping in Inner Mongolia.Methods The clinical M. tuberculosis strains isolated from confirmed tuberculosis patients, including the patients’ epidemiological and clinical information, were collected from Inner Mongolia Institute for Tuberculosis Control and Prevention and analyzed by the way of multi-Locus VNTR analysis(MLVA) with 22 VNTR loci. The polymorphism of different 22 VNTR loci were analyzed by the Hunter-Gaston Index with Bionumerics 5.0 software.Results Of all the 372 strains, 308 genotypes with 22 VNTR loci were detected, including 47 genotype clustering and 261 distinct genotypes. The clustering rate of genotype was 17.20%. In addition, the 22 loci had different polymorphisms, VNTR3820(HGDI 0.838) locus had highest discriminatory power, whereas MIRU23(HGDI 0.068) and MIRU27(HGDI 0.083) loci had lower discriminatory power. With the increase of the number of VNTR loci, the discriminatory power of VNTR analysis for M. tuberculosis strains was improved.Conclusion Gene polymorphism of clinical M. tuberculosis strains was observed in Inner Mongolia, and the different loci had distinct discriminatory power in VNTR analysis.
内蒙古地区结核分枝杆菌串联重复序列基因多态性分析
苏云开1, 余琴2,3, 吕冰2, 马岩3, 连璐璐2,3, 杨晓敏1, 董海燕2, 刘耀1, 赵秀芹2, 吴移谋3, 万康林2,3
1. 内蒙古自治区结核病防治所, 内蒙古 呼和浩特 010010;
2. 中国疾病预防控制中心传染病预防控制所, 传染病预防控制国家重点实验室;
3. 南华大学病原生物学研究所
2. 中国疾病预防控制中心传染病预防控制所, 传染病预防控制国家重点实验室;
3. 南华大学病原生物学研究所
摘要
目的 了解内蒙古地区结核分枝杆菌临床分离株的串联重复序列(variable number tandem repeat,VNTR)基因多态性和VNTR基因型构成,及不同VNTR位点在该地区结核分枝杆菌基因分型中的应用。 方法 从内蒙古自治区结核病防治研究所收集临床分离的结核分枝杆菌菌株及其病例背景资料,采用多位点串联重复序列分析(MLVA)对收集的菌株进行22个VNTR 位点分析,计算Hunter-Gaston 指数(HGDI),分析各个位点的分辨率, 同时用BioNumerics 软件对VNTR结果进行聚类分析。且统计学分析民族与主要基因型间的关系。 结果 372株菌共分为308个基因型,47簇,261个独特基因型,成簇率为17.20%。22个VNTR位点的基因多态性存在较大的差异,位点VNTR3820(HGDI 0.838)的分型分辨能力最高,MIRU23(HGDI 0.068)和MIRU27(HGDI 0.083)分型分辨能力较差。随着VNTR 位点的增加,分型的分辨能力也有所提高。Ⅰ群基因型菌株与民族易感性的分析表明,汉族与蒙古族间Ⅰ群基因型菌株分布差异无统计学意义(χ2=0.337, P=0.561>0.05)。 结论 内蒙古地区结核分枝杆菌临床分离株基因具有多态性,不同VNTR位点在内蒙古地区结核分枝杆菌中具有不同的分辨能力。且Ⅰ群基因型为该地区主要流行株,而Ⅰ群基因型菌株与民族易感性间无关联。
内容大纲
-
1 材料与方法
- 1.1 菌株
- 1.2 主要试剂
- 1.3 DNA 提取
- 1.4 多位点串联重复序列分析(MLVA)
- 1.4.1 聚合酶链反应(PCR)反应体系
- 1.4.2 凝胶电泳分析
- 1.5 统计学处理
- 2.1 聚类分析
- 2.2 22位点多态性分析
- 2.3 基因群与内蒙古地区主要民族易感性分析
随着分子生物学技术的发展,结核分枝杆菌分子分型的方法在探讨结核病的传播特征、追踪传染源、监测可疑暴发及研究遗传相关性方面具有重要的科学实用性[2]。其中,基于分枝杆菌数目可变串联重复序列(mycobacterial interspersed repetitive units-variable number tandem repeat, MIRU-VNTR)的结核分枝杆菌分型方法,因其分辨率、敏感性、特异性和重复性均较高,且结果数字化,易于比较等优势,成为结核病流行病学研究中应用较为广泛的方法之一。目前,由于世界各地结核分枝杆菌基因型的差别较大,因此MIRU-VNTR分型位点的选择尚未有一致的标准。我们根据中国目前已有的文献报道,选取22位点,对内蒙古地区结核分枝杆菌临床分离株进行基因分型,旨在对该地区结核病分子流行病学进行研究,从而为该地区结核病传染源的追溯、传播机制及菌株遗传学关系等的研究提供理论基础。
1 材料与方法
1.1 菌株372株结核分枝杆菌来源于2011年内蒙古自治区结核病防治研究所临床病例痰标本。其中316个新发病例,56个复治病例;男性245例,女性124例,3例性别不详。汉族282例,蒙古族84例,回族4例,维吾尔族1例,满族1例。结核分枝杆菌标准菌株H37Rv由中国疾病预防控制中心(CDC)传染病预防控制所结核病实验室提供。
1.2 主要试剂
2×PCR Taq MasterMix和100 bp Ladder购自北京康为世纪生物科技有限公司;引物合成由上海生物工程技术服务有限公司进行;L-J培养基由中国CDC传染病预防控制所结核病实验室制备提供。
1.3 DNA 提取
取一菌环L-J培养基斜面上生长的结核分枝杆菌,置于400 μl TE 缓冲液中,沸水浴20 min,12 000 r/min 离心5 min,取上清置于-20 ℃保存备用。
1.4 多位点串联重复序列分析(MLVA)
1.4.1 聚合酶链反应(PCR)反应体系
选取22个MIRU-VNTR位点,见表1。采用12 μl反应体系:上下游引物(5 μmol/L)各1 μl,DNA模板2 μl,2×PCR Taq MasterMix 6 μl,2 μl RNase-Free水。PCR反应条件为94 ℃ 10 min, 94 ℃ 30 s, 62 ℃ 30 s,72 ℃ 45 s,35个循环,终止延伸10 min。PCR反应引物参考文献[3],具体见表1。
1.4.2 凝胶电泳分析
PCR扩增产物经2.0%的琼脂糖凝胶电泳,凝胶成像系统进行拍照,Image Lab 软件对电泳条带进行分析,计算重复片段数。
1.5 统计学处理
将所有菌株的VNTR位点进行Excel录入,经BioNumerics软件进行聚类。计算各VNTR位点的分辨指数HGDI,公式如下:
HGDI=1-1/N(N-1)∑ s j=1 nj(nj-1)
式中N为菌株总数,S是总基因型数,nj代表第j类基因型的菌株数[4]。
分析基因型完全相同的菌株,定义为一簇。成簇率的计算如下:(nc-c)/n,nc代表成簇的菌株数,c为成簇数,n为总菌株数。
2 结果
2.1 聚类分析 根据MIRU-VNTR分型结果,BioNumerics进行聚类,372株菌共表现出308个基因型,其中261株菌表现为独特基因型,111株表现为47簇,成簇率为17.20%,具体聚类分析见图1。
2.2 22位点多态性分析
利用Hunter-Gaston(HGDI)指数评价各VNTR位点的基因多态性,根据 文献报道[5],基于HGDI 的不同,VNTR位点可分为高分辨率(HGDI>0.6)、中分辨率(0.3<HGDI<0.6)和低分辨率(HGDI<0.3)。 在本实验中,VNTR3820、 Qub11b、Qub18、 Qub11a、 Mtub21 和 Qub26等6个位点的HGDI均大于0.6,属于高分辨率位点;MIRU26、Mtub04、ETRA、ETRE、MIRU10、Mtub30、MIRU39和Qub4156c等8个位点的HGDI在0.3~0.6之间,属于中分辨率位点,而剩下的则分辨率较低。22位点累计HGDI为0.9902。具体各位点HGDI值见表2。
2.3 基因群与内蒙古地区主要民族易感性分析
根据聚类分析,372株结核 分枝杆菌主要 分为5大基
表1 各重复位点在H37Rv中的重复次数、重复片段大小及其引物序列
Table 1 Primer sequence, size of repeats and number of repeats in M. tuberculosis H37Rv
| 序号 | 名称 | 引物序列(5′~3′) | 重复片段大小 (bp) | 重复次数 | 扩增片段大小 (bp) |
| 1 | ETRA | L) ATT TCG ATC GGG ATG TTG AT R) TCG GTC CCA TCA CCT TCT TA | 75 | 3 | 397 |
| 2 | ETRB | L) GCG AAC ACC AGG ACA GCA TCA TG R) GGC ATG CCG GTG ATC GAG TGG | 57 | 3 | 292 |
| 3 | ETRC | L) GAC TTC AAT GCG TTG TTG GA R) GTC TTG ACC TCC ACG AGT GC | 58 | 4 | 346 |
| 4 | ETRD | L) GCG CGA GAG CCC GAA CTG C R) GCG CAG CAG AAA CGT CAG C | 77 | 3 | 330 |
| 5 | ETRE | L) ACT GAT TGG CTT CAT ACG GCT TTA R)GTG CCG ACG TGG TCT TGA T | 53 | 3 | 651 |
| 6 | MIRU10 | L) GTT CTT GAC CAA CTG AGT CGT CC R) GCC ACC TTG GTG ATC AGC TAC CT | 53 | 3 | 643 |
| 7 | MIRU16 | L) TCG GTG ATC GGG TCC AGT CCA AGT A R) CCC GTC GTG CAG CCC TGG TAC | 53 | 2 | 671 |
| 8 | MIRU23 | L) CAG CGA AAC GAA CTG TGC TAT CAC R) CGT GTC CGA GCA GAA AAG GGT AT | 53 | 6 | 873 |
| 9 | MIRU26 | L) CCC GCC TTC GAA ACG TCG CT R) TGG ACA TAG GCG ACC AGG CGA ATA | 51 | 3 | 613 |
| 10 | MIRU27 | L) TCG AAA GCC TCT GCG TGC CAG TAA R) GCG ATG TGA GCG TGC CAC TCA A | 53 | 3 | 657 |
| 11 | MIRU39 | L) CGC ATC GAC AAA CTG GAG CCA AAC R) CGG AAA CGT CTA CGC CCC ACA CAT | 53 | 2 | 646 |
| 12 | MIRU40 | L) GGG TTG CTG GAT GAC AAC GTG T R) GGG TGA TCT CGG CGA AAT CAG ATA | 54 | 1 | 407 |
| 13 | Mtub21 | L) AGA TCC CAG TTG TCG TCG TC R) CAA CAT CGC CTG GTT CTG TA | 57 | 2 | 206 |
| 14 | Mtub30 | L) AGT CAC CTT TCC TAC CAC TCG TAA C R) ATT AGT AGG GCA CTA GCA CCT CAA G | 58 | 2 | 319 |
| 15 | Mtub39 | L) AAT CAC GGT AAC TTG GGT TGT TT R) GAT GCA TGT TCG ACC CGT AG | 58 | 6 | 515 |
| 16 | Qub11a | L) CCC ATC CCG CTT AGC ACA TTC GTA R) TTC AGG GGG GAT CCG GGA | 69 | 3 | 305 |
| 17 | Qub11b | L) CGT AAG GGG GAT GCG GGA AAT AGG R) CGA AGT GAA TGG TGG CAT | 69 | 5 | 412 |
| 18 | Qub4156c | L) TGG TCG CTA CGC ATC GTG TCG GCC CGT R) TAC CAC CCG GGC AGT TTA C | 59 | 3 | 224 |
| 19 | Qub18 | L) ATC GTC AGC TGC GGA ATA GT R) AAT ACC GGG GAT ATC GGT TC | 78 | 6 | 621 |
| 20 | Qub26 | L) AAC GCT CAG CTG TCG GAT R) GGC CAG GTC CTT CCC GAT | 111 | 5 | 708 |
| 21 | Mtub04 | L) GTC CAG GTT GCA AGA GAT GG R) GGC ATC CTC AAC AAC GG TAG | 51 | 3 | 269 |
| 22 | VNTR3820 | L) TGC GCG GTG AAT GAG ACG R) ACC TTC ATC CTT GGC GAC | 57 | 3 | 409 |
因群(Ⅰ群257株,Ⅱ群46株,Ⅲ群25株,Ⅳ群38株,Ⅴ群10株,分别占69.09%、12.37%、4.03%、10.22%和2.69%),其他为单个独立菌株。其中282例为汉族,84例为蒙古族,且Ⅰ群257株,因此分析两个主要民族与Ⅰ群基因型菌株的易感性,χ2检验显示,两个民族间的Ⅰ群基因型菌株与非Ⅰ群基因型菌株差异无统计学意义(P>0.05),见表3。
3 讨论
内蒙古自治区在中国属于结核病严重流行地区之一。早在1990年全国结核病流行病学调查显示,内蒙古地区结核病的流行率仅次于西藏和四川,居全国第3位,且高于1979年的调查数据[6]。因此,对该地区结核分枝杆菌分子流行病学开展研究具有重要的意义。
图1 内蒙古自治区372株结核分枝杆菌BioNumerics聚类分析图
Figure 1 BioNumerics analysis of 372 M. tuberculosis isolates from Inner Mongolia
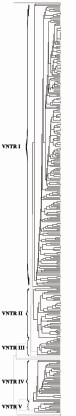
表2 22个VNTR位点在372株结核分枝杆菌中不同的Hunter-Gaston 指数
Table 2 HGDI scores of 22 MIRU-VNTR loci for 372 M. tuberculosis isolates
| 序号 | 位点 | HGDI | 序号 | 位点 | HGDI |
| 1 | VNTR3820 | 0.838 | 13 | MIRU39 | 0.327 |
| 2 | Qub11b | 0.756 | 14 | Qub4156c | 0.316 |
| 3 | Qub18 | 0.706 | 15 | MIRU40 | 0.299 |
| 4 | Qub11a | 0.663 | 16 | Mtub39 | 0.285 |
| 5 | Mtub21 | 0.655 | 17 | MIRU16 | 0.204 |
| 6 | Qub26 | 0.624 | 18 | ETRB | 0.194 |
| 7 | MIRU26 | 0.548 | 19 | ETRD | 0.134 |
| 8 | Mtub04 | 0.461 | 20 | ETRC | 0.127 |
| 9 | ETRA | 0.425 | 21 | MIRU27 | 0.083 |
| 10 | ETRE | 0.402 | 22 | MIRU23 | 0.068 |
| 11 | MIRU10 | 0.388 | 23 | cumulative HGDI | 0.9902 |
| 12 | Mtub30 | 0.334 |
表3 Ⅰ群基因型菌株与主要民族间易感性分析
Table 3 Correlation between VNTR Ⅰgenotype and ethnic groups
| 民族 | Ⅰ群基因型 菌株数 | 其他基因型 菌株数 | 合计 (菌株数) |
| 汉族 | 192 | 90 | 282 |
| 蒙古族 | 60 | 24 | 84 |
| 合计 | 252 | 114 | 366 |
通过MIRU-VNTR分型显示,本研究372株结核分枝杆菌共表现出308个基因型,其基因多态性明显,其中261株菌表现为独特基因型,111株表现为47簇,成簇率为17.20%。较spologotyping(另文发表)结果比较,分型结果明显较好。内蒙古作为中国少数民族聚居最多的地区,其结核病分子流行病学的基因多态性表现对于民族易感性的研究具有一定意义。
MIRU-VNTR分型是结核分枝杆菌基因分型应用较广泛的方法之一,但在不同地区,由于结核分枝杆菌基因型的差别,其VNTR位点的选择亦具有不同的差异性。常用的MIRU-VNTR分型包括12位点、15位点和24位点组合,其分辨率因位点组合的不同而不同 。据最新文献报道 ,日本、尼日利亚和北俄罗斯分别采用JATA-VNTR、 24 VNTR和 15 VNTR(12 VNTR加3高变位点)组合来分析各自地区的结核分枝杆菌及其北京家族菌株,其分辨率均较高。由此可见,各地区VNTR分型位点的选择各有差异,其依据为该地区结核分枝杆菌 流行的主要基因型。在本实验中,选取的22位点是在先前其他省份的研究基础上 ,挑选多态性较高的位点,对菌株进行分析。累积分辨率为0.9902。
基于统计学数据,内蒙古自治区2007年人口调查显示78%的人口为汉族,18%的为蒙古族,且蒙古族的比例在逐年上升[14]。在本研究中,蒙古族和汉族人口的比例分别为22.58%、75.81%,符合人口基本状况,因此统计学分析是有意义的。而其他少数民族由于人口比例较低,不满足统计学要求,而未纳入统计分析中。有关Ⅰ群基因型菌株易感性和主要民族相关性分析显示,内蒙古地区结核分枝杆菌Ⅰ群基因型菌株的感染与汉族和蒙古族无关,同时由于菌株来源在各区分布不一致,因此未能有效讨论内蒙古不同地理与结核病的流行状况相关性。
综上所述,对于内蒙古地区结核分枝杆菌的基因型构成及流行株有了初步了解,这对今后的结核病防控工作有一定的指导意义。另外,还需对该地区北京家族的流行及其耐药等多方面进行深入研究探讨,才能更好的提高结核病的防控水平。
参考文献
[1] WHO(2010) Global tuberculosis control 2010:epidemiology, strategy, financing. Geneva:WHO, Available://www.who.int/tb.en.
[2] Barnes PF, Cave MD. Molecular epidemiology of tuberculosis[J]. N Engl J Med, 2003, 349:2364.
[3] Supply P, Allix C, Lesjean S, et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unitvariablenumber tandem repeat typing of Mycobacterium tuberculosis [J]. J Clin Microbiol, 2006, 44:4498-4510.
[4] Hunter PR, Gaston MA. Numerical index of the discriminatory ability of typing systems:an application of Simpson’s index of diversity[J]. Clin Microbiol, 1988, 26:2465-2466.
[5] Mokrousov I, Narvskaya O, Limeschenko E, et al. Analysis of the allelic diversity of the mycobacterial interspersed repetitive units in Mycobacterium tuberculosis strains of the Beijing family:practical implications and evolutionary considerations [J]. J Clin Microbiol, 2004, 42:2438-2444.
[6] The national tuberculosis epidemiology sampling technology steering group. The third national tuberculosis epidemiology sampling investigation report [J]. Chinese Journal of Tuberculosis and Respiratory Diseases, 1992, 15:69-71.(in Chinese)
全国结核病流行病学抽样调查技术指导组. 第三次全国结核病流行病学抽样调查报告[J].中华结核和呼吸杂志, 1992, 15:69-71.
[7] Liu Q, Yang D, Xu W, et al. Molecular typing of Mycobacterium tuberculosis isolates circulating in Jiangsu province, China [J]. BMC Infect Dis, 2011, 11:288.
[8] Yuhki N, Yoshito I, Eri H, et al. Molecular Genotyping of Mycobacterium tuberculosis in Mie Prefecture, Japan, Using Variable Numbers of Tandem Repeats Analysis [J]. Jpn J Infect Dis, 2012, 65:341-344.
[9] Lawson L, Zhang J, Gomgnimbou MK, et al. A molecular epidemiological and genetic diversity study of tuberculosis in Ibadan, Nnewi and Abuja, Nigeria [J]. PLoS One, 2012, 7:e38409.
[10] Mokrousov I, Vyazovaya A, Otten T, et al. Mycobacterium tuberculosis Population in Northwestern Russia:An Update from RussianEU/Latvian Border Region [J]. PLoS One, 2012, 7(7):e41318.
[11] Chai LQ, Li WM, Li L, et al. Study on the genotype of Mycobacterium tuberculosis isolates from hospitals in Tianjin[J]. Chinese Journal of Epidemiology, 2007, 28(8):785-788.(in Chinese)
柴利泉, 李卫民, 李丽, 等.天津地区临床分离结核分枝杆菌分型的初步研究[J]. 中华流行病学杂志, 2007, 28(8):785-788.
[12] Dong H, Liu Z, Lv B, et al. Spoligotypes of Mycobacterium tuberculosis from different Provinces of China [J]. J Clin Microbiol, 2010, 48:4102-4106.
[13] Wang J, Liu Y, Zhang CL, et al. Genotypes and characteristics of clustering and drug susceptibility of Mycobacterium tuberculosis isolates collected in Heilongjiang province, China [J]. J Clin Microbiol, 2011, 49:1354-1362.
[14] Zhao LC. Inner Mongolia minority population quantity change, causes and countermeasures[M]. Hohhot city:Inner Mongolia Normal University, 2010.(in Chinese)
[2] Barnes PF, Cave MD. Molecular epidemiology of tuberculosis[J]. N Engl J Med, 2003, 349:2364.
[3] Supply P, Allix C, Lesjean S, et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unitvariablenumber tandem repeat typing of Mycobacterium tuberculosis [J]. J Clin Microbiol, 2006, 44:4498-4510.
[4] Hunter PR, Gaston MA. Numerical index of the discriminatory ability of typing systems:an application of Simpson’s index of diversity[J]. Clin Microbiol, 1988, 26:2465-2466.
[5] Mokrousov I, Narvskaya O, Limeschenko E, et al. Analysis of the allelic diversity of the mycobacterial interspersed repetitive units in Mycobacterium tuberculosis strains of the Beijing family:practical implications and evolutionary considerations [J]. J Clin Microbiol, 2004, 42:2438-2444.
[6] The national tuberculosis epidemiology sampling technology steering group. The third national tuberculosis epidemiology sampling investigation report [J]. Chinese Journal of Tuberculosis and Respiratory Diseases, 1992, 15:69-71.(in Chinese)
全国结核病流行病学抽样调查技术指导组. 第三次全国结核病流行病学抽样调查报告[J].中华结核和呼吸杂志, 1992, 15:69-71.
[7] Liu Q, Yang D, Xu W, et al. Molecular typing of Mycobacterium tuberculosis isolates circulating in Jiangsu province, China [J]. BMC Infect Dis, 2011, 11:288.
[8] Yuhki N, Yoshito I, Eri H, et al. Molecular Genotyping of Mycobacterium tuberculosis in Mie Prefecture, Japan, Using Variable Numbers of Tandem Repeats Analysis [J]. Jpn J Infect Dis, 2012, 65:341-344.
[9] Lawson L, Zhang J, Gomgnimbou MK, et al. A molecular epidemiological and genetic diversity study of tuberculosis in Ibadan, Nnewi and Abuja, Nigeria [J]. PLoS One, 2012, 7:e38409.
[10] Mokrousov I, Vyazovaya A, Otten T, et al. Mycobacterium tuberculosis Population in Northwestern Russia:An Update from RussianEU/Latvian Border Region [J]. PLoS One, 2012, 7(7):e41318.
[11] Chai LQ, Li WM, Li L, et al. Study on the genotype of Mycobacterium tuberculosis isolates from hospitals in Tianjin[J]. Chinese Journal of Epidemiology, 2007, 28(8):785-788.(in Chinese)
柴利泉, 李卫民, 李丽, 等.天津地区临床分离结核分枝杆菌分型的初步研究[J]. 中华流行病学杂志, 2007, 28(8):785-788.
[12] Dong H, Liu Z, Lv B, et al. Spoligotypes of Mycobacterium tuberculosis from different Provinces of China [J]. J Clin Microbiol, 2010, 48:4102-4106.
[13] Wang J, Liu Y, Zhang CL, et al. Genotypes and characteristics of clustering and drug susceptibility of Mycobacterium tuberculosis isolates collected in Heilongjiang province, China [J]. J Clin Microbiol, 2011, 49:1354-1362.
[14] Zhao LC. Inner Mongolia minority population quantity change, causes and countermeasures[M]. Hohhot city:Inner Mongolia Normal University, 2010.(in Chinese)