1. Yunnan Provincial Institute for Endemic Disease Control and Prevention, Dali 671000, Yunnan, China;
2. School of public health, Dali University, Dali 671000, Yunnan, China
Multiple-locus variable-number tandem-repeat analysis on Yersinia pestis in Yunnan
ZHU Jun-jie1,2, WANG Peng1, ZHANG Rong2, HAI Rong3, LIANG Ying3, SONG Zhi-zhong1
Abstract
Objective To identify genotypes of Yersinia pestis in Yunnan by using Multiple-locus variable-number tandem-repeat (VNTR) analysis (MLVA). Methods By using polymerase chain reaction (PCR) and capillary electrophoresis, the genotypes of 158 Yersinia pestis strains isolated in Yunnan province were analyzed, and the data were processed with BioNumerics software. Results Five VNTR sites can be used to identify the genotypes of Yersinia pestis in Yunnan. The clustering of genotype showed that these strains could be classified into 2 groups, 3 clusters and 5 genotypes. The wild rodent strains belonged to cluster A, the stains isolated in Yulong belonged to cluster B and commensal rodent strains belonged to cluster C. The Results were consistent with traditional ecological classification. Conclusion The wild rodent strains, commensal rodent strains and Yulong strains isolated in Yunnan belonged to different gene clusters, but Yulong strains and wild rodent strains were in one group. The clustering analysis showed good relationship between the genotypes of Yersinia pestis and geographic location.
Keywords:
Yersinia pestis
multiple-locus variable-number tandem-repeat analysis
genotype
ecological classification
云南省鼠疫耶尔森菌多位点可变数目串联重复序列分析
朱俊洁1,2, 王鹏1, 张蓉2, 海荣3, 梁莹3, 宋志忠1
1. 云南省地方病防治所云南省鼠疫防控技术重点实验室, 云南 大理 671000;
2. 大理学院公共卫生学院, 云南 大理 671000;
3. 中国疾病预防控制中心传染病预防控制所鼠疫室
2. 大理学院公共卫生学院, 云南 大理 671000;
3. 中国疾病预防控制中心传染病预防控制所鼠疫室
摘要
目的 通过多位点可变数目串联重复序列分析(multiple-locus variable-number tandem-repeat analysis,MLVA)方法,对云南省鼠疫菌株进行基因分型。 方法 采用聚合酶链反应(PCR)和毛细管电泳,通过BioNumerics软件进行处理,分析云南省158株鼠疫耶尔森菌基因型。 结果 选取鼠疫菌的14个VNTR位点,5个VNTR位点对云南省鼠疫菌具有多态性分析意义,利用这5个位点对云南菌株进行分析,云南158株鼠疫菌可分为2个群,3个簇,5个基因型,野鼠鼠疫菌属于A簇,丽江玉龙鼠疫菌属于B簇,家鼠鼠疫菌属于C簇,与传统的生态分型结果吻合。 结论 本次试验筛选的5个位点可以用于云南省鼠疫菌的分型,云南家鼠鼠疫菌、野鼠鼠疫菌及玉龙鼠疫菌分属不同的基因簇,玉龙鼠疫菌与野鼠鼠疫菌同属一个群,聚类结果与实际的地理相关性很好。
内容大纲
-
1 材料与方法
- 1.1 菌株来源及DNA制备
- 1.2 VNTR位点选择
- 1.3 聚合酶链反应(poly chain reaction,PCR)扩增及条件
- 1.4 毛细管电泳
- 1.5 计算分辨指数及聚类分析
- 3.1 云南省鼠疫菌VNTR特点分析
- 3.2 云南省鼠疫菌MLVA分型特征及其流行病学意义
分析云南省多年分离的鼠疫菌株,对研究菌株遗传特征和疫源地之间的联系及其对人群的危害具有重要的意义。MLVA方法是细菌众多基因分型方法中的一种,被国外研究者广泛应用 ,在国内有研究者利用国外已公布的部分VNTR位点对中国鼠疫菌进行分子分型 。本研究通过对13株已公布鼠疫菌的全基因进行比对,筛选了14个VNTR位点(其中12个位点是全新的位点,另2个是已公布使用过的位点[7]),首次对云南省158株鼠疫菌进行MLVA分子分型,一方面评价这些位点对云南菌株的分型能力,另一方面探究云南鼠疫菌株之间遗传关系,了解他们之间的内在联系,弄清云南省鼠疫疫源地存在与活动的流行规律, 从而为云南省的鼠疫防控工作提供更有力的科学依据,结果如下。
1 材料与方法
1.1 菌株来源及DNA制备 云南省分离的158株鼠疫菌(其中云南家鼠鼠疫菌株135株、野鼠鼠疫菌株16株、丽江玉龙菌株7株)及疫苗株EV76,由云南省地方病防治所云南省鼠疫防控技术重点实验室提供,见表1。利用DNA提取试剂盒(QIAGEN,德国)提取鼠疫菌DNA,置-20 ℃保存。
表1 实验所用菌株信息
Table 1 Information of strains used in study
| 分离地点 | 数目 | 分离宿主 | 分离时间(年.月) | ||
| 鼠 | 蚤 | 患者 | |||
| 大理州(1) | 16 | 10 | 6 | 0 | 1954.11 1990.03 |
| 丽江市(2) | 7 | 5 | 2 | 0 | 2006.11 2009.05 |
| 保山市<sup>(3) | 10 | 5 | 5 | 0 | 1983.09 1994.11 |
| 德宏州(3) | 42 | 20 | 18 | 4 | 1964.10 2007.11 |
| 红河州(3) | 15 | 18 | 0 | 0 | 1996.12 2002.09 |
| 昆明市(3) | 1 | 1 | 0 | 0 | 1997.12 |
| 临沧市(3) | 32 | 15 | 8 | 9 | 1990.12 2001.11 |
| 普洱市(3) | 15 | 8 | 6 | 1 | 1992.01 2001.05 |
| 思茅市(3) | 2 | 2 | 0 | 0 | 1995.11 |
| 文山州(3) | 11 | 5 | 2 | 4 | 1996.12 2002.07 |
| 西双版纳(3) | 6 | 6 | 0 | 0 | 1992.02 1999.04 |
| 玉溪市(3) | 1 | 1 | 0 | 0 | 1992.04 |
1.2 VNTR位点选择
VNTR位点是根据已公布的13株鼠疫菌序列比对筛选得到的串联重复基因位点(http://minisatellites.u-psud.fr),由中国疾病预防控制中心传染病预防控制所鼠疫室提供,见表2。根据位点序列设计扩增引物,并用FAM、HEX、ROX三种不同的荧光进行标记,引物由上海辉睿生物科技公司合成。
表2 MLVA位点引物列表及其核心序列长度
Table 2 Primer sequence of MLVA locus and size of core sequence
| 位点 | 引物(5′~3′) | 核心序列长度(bp) | 理论产物长度(bp)(1) | 重复数(1) |
| MLV1 | F:CCA AGG CCC GTT TCC GGG TG R:CCA ACC AAC TGC GCG CCT CT | 143 | 1367 | 4.9 |
| MLV2 | F:CCT GCG TCT GGG CAA ATG GC R:TCG CCG TAA GGC CCA TCA ACC T | 123 | 877 | 3.0 |
| MLV3 | F:TCG CCC TGT GCA GTT GCT CG R:GGC AGT GAA GCG GCG ACT GT | 113 | 1200 | 4.7 |
| MLV4 | F:TCC CGG AGA AGG GGA ACA GCG R:GGC TGA CAC CAG AGC AAC AAC A | 69 | 760 | 3.6 |
| MLV5 | F:AGC ATC CAT CAG GTG AAT CCC GC R:AGC GCC GTT GTT CTG CCA GC | 18 | 251 | 3.5 |
| MLV6 | F:TCA CCT TCA CTT ATC GAC AGC AGC A R:AGC GCC GTT GTT CTG CCA GC | 15 | 240 | 4.2 |
| MLV7 | F:AGA CAC TGA CCG CGC CGA TG R:TTT GCG CTC TTC CCC CTG CC | 18 | 224 | 2.5 |
| MLV8 | F:TCT GGC TTT GGC TTC TCC TTG CC R:AAG GAC GGA TCG CCT CTA CCA G | 10 | 362 | 2.5 |
| MLV9 | F:TGG CGG CGG AGT TTT TCG CT R:CCC CGC AAA ACG TGA CTG TCG | 15 | 376 | 5.0 |
| MLV11 | F:AGC GTT GAC AAC CCA ATA TGT GGA A R:AGG GCA GTC GGG TGG ATG CA | 18 | 449 | 3.0 |
| MLV12 | F:TGC AGG AAG GCG GCA ACT GAG R:GTG ACT GCT GCT AAT TCG GGC CA | 21 | 334 | 2.0 |
| MLV14 | F:CCC GTC ACC GAC TGG GAA TGC R:CGC AAA TCA TGT CGG CCG CG | 21 | 444 | 2.7 |
| M52 | F:GTG GCC TAA CCC GTT TTA CCG GTG TAG C R:GAG TCC TGA TTC GTG ATT GAC AAA ACC GC | 15 | 202 | 4.0 |
| M59 | F:GAT AAT GGC GGT AGC CGG AAT CTG ATA ATC ATC R:GCC AAC TCA CCT TTT CTG GCG GCT AAG C | 17 | 296 | 8.0 |
1.3 聚合酶链反应(poly chain reaction,PCR)扩增及条件
制备的鼠疫DNA用去离子水30倍稀释为 PCR模板。采用总的体积25 μl的多重PCR反应体系,PCR反应条件:94 ℃预变性5 min,94 ℃变性45 s,60 ℃退火45 s,72 ℃延伸90 s,35个循环,72 ℃延伸5 min。
1.4 毛细管电泳
PCR产物进行毛细管电泳分析,通过PCR产物大小计算出重复数。
1.5 计算分辨指数及聚类分析
通过BioNumerics6.6软件采用非加权组平均法(unweighted pair-group method with arithmetic means,UPGMA)对各位点重复数进行数据分析,得到各位点的分辨指数(Simpsons Index of Diversity),绘制聚类图和最小生成树。
2 结果
2.1 各位点分辨力 各位点的分辨力差别较大。以实验中的159株鼠疫菌为基础计算的M52位点的分辨指数为0.249,该位点将159株菌分为2组,各组的重复数分别为3.7(135株)、2.7(24株)。MLV11位点的分辨指数为0.012,仅1株菌1393号重复数为3.4,其他均为2.7。另外,有8个位点的分辨指数为0,其他5个位点(M52、M59、MLV7、MLV8、MLV4)有一定的分辨力,结果见表3。
表3 159株云南省鼠疫菌在14个位点的重复次数的分辨指数计算结果
Table 3 Index of diversity calculation results of repetitions at 14 VNTR locus in 159 Yersinia pestis stains in Yunnan
| 位点名称 | 指数 | 组数 | 重复数 | 菌株数 |
| M52 | 0.249 | 2 | 3.7 2.7 | 135 24 |
| M59 | 0.240 | 2 | 3.7 2.7 | 135 24 |
| MLV7 | 0.240 | 2 | 2.4 5.4 | 136 23 |
| MLV8 | 0.097 | 3 | 2.3 3.3 5.3 | 2 147 10 |
| MLV4 | 0.085 | 2 | 2.5 3.5 | 11 148 |
| MLV11 | 0.012 | 2 | 2.7 3.4 | 158 1 |
| MLV1 | 0 | 1 | 5.0 | 159 |
| MLV2 | 0 | 1 | 3.0 | 159 |
| MLV3 | 0 | 1 | 4.7 | 159 |
| MLV5 | 0 | 1 | 3.4 | 159 |
| MLV6 | 0 | 1 | 4.2 | 159 |
| MLV9 | 0 | 1 | 4.9 | 159 |
| MLV12 | 0 | 1 | 1.8 | 159 |
| MLV14 | 0 | 1 | 2.6 | 159 |
2.2 VNTR基因分型
利用M52、M59、MLV7、MLV8、MLV4这5个位点重复数,采用软件BioNumerics6.6对云南地区的159株鼠疫菌(包括EV菌)进行聚类分析及最小生成树绘制,结果见图1、图2,159株鼠疫菌(包括EV菌)分为两大群,三个簇,5个基因型即1~5型。Ⅰ群野鼠鼠疫群包括大理剑川野鼠菌株和丽江玉龙鼠疫菌株,分为A簇、B簇。A簇被分为两个基因型,与云南省生态型-滇西纵谷型吻合较好,7株丽江玉龙菌株为B簇,为一个独立的生态型,与A簇的剑川鼠疫菌株5个位点中有2个位点差异。Ⅱ群中家鼠鼠疫(为包括EV菌)为C簇,分为两个基因型,与云南省生态型-滇闽居民型吻合较好。
3 讨论
3.1 云南省鼠疫菌VNTR特点分析本次实验所选取的VNTR位点核心序列长度差别较大,Klevytska 等[3]研究表明,核心序列越小的位点,等 位基因数越大,对菌株的分辨能力越强。但是在本次研究中没有明显地显示这种规律,如MLV7和MLV8的核心序列长度分别为18 bp和10 bp,但它们对实验菌株的分辨指数分别为0.240和0.097。所选取的14个位点,只有M52和M59有相关研究报道曾用于鼠疫菌的分型[7],其他均为自行设计的新位点,其中8个位点在云南省鼠疫菌上比较稳定,
图1 159株菌的最小生成树
Figure 1 Minimum spanning tree of 159 strains
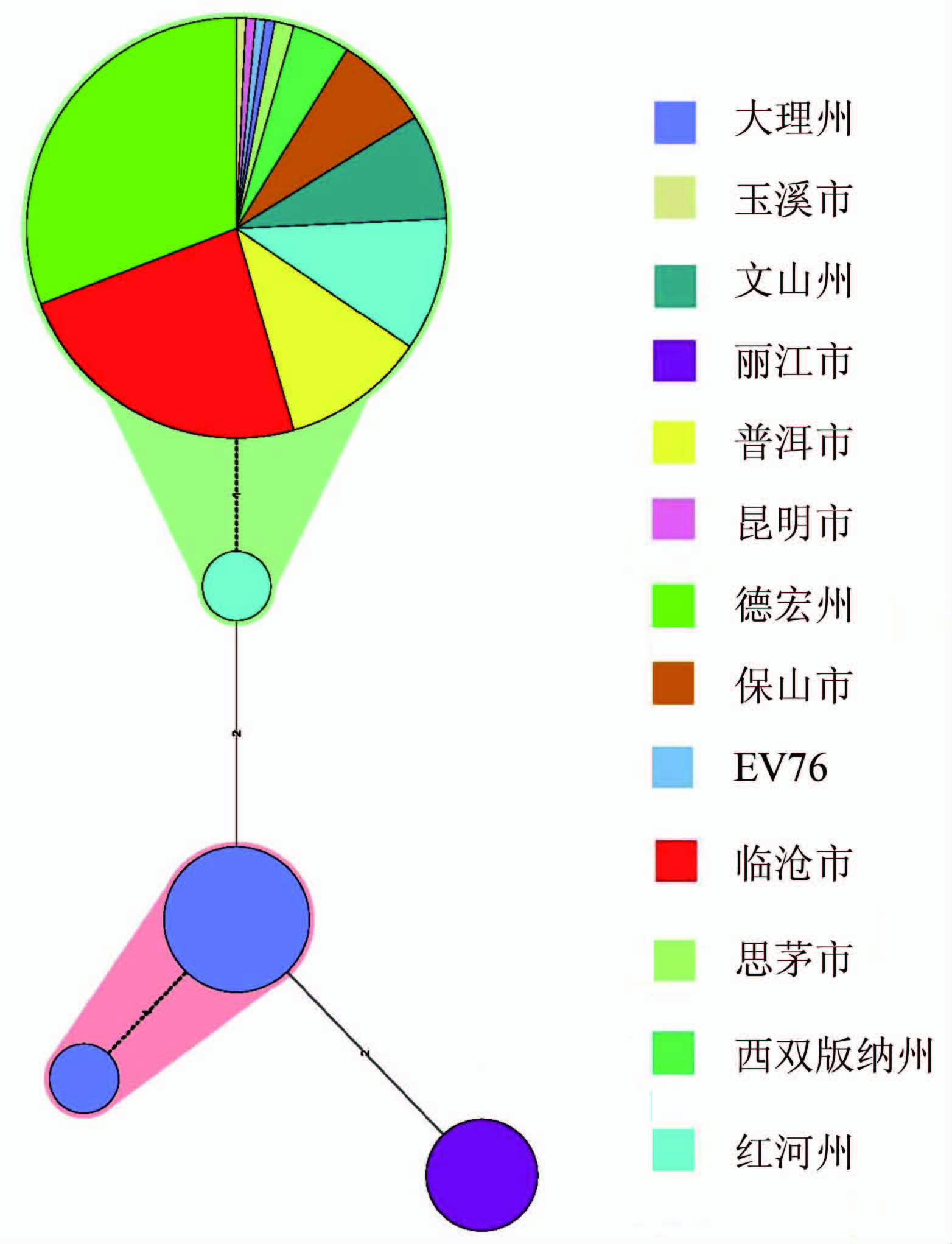
实线表示2个重复数的差异,虚线表示1个重复数的差异,同一底色表示分为同一个簇,圆盘大小由菌株数量而定。
图2 159株菌MLVA基因分型的聚类分析
Figure 2 Cluster analysis of 159 strains through MLVA genotyping

基本上不存在多态性。大部分菌株在其他有分辨能力的5个位点上核心序列重复数只有2种,其多样性也较低。
3.2 云南省鼠疫菌MLVA分型特征及其流行病学意义
云南野鼠菌株(大理剑川)大部分属于一个MLVA基因型(A簇2型),鼠疫菌332号菌株被分为一个独立的MLVA型即1型,与其他的野鼠菌株存在一个位点的差异,该菌株分离自野鼠疫源地,但其宿主是黄胸鼠,且分离于1954年,这也提示随着时间和生存环境的改变,鼠疫菌基因可能存在一定程度的突变。
玉龙鼠疫菌属于MLVA基因Ⅰ群中的B基因簇,且所有菌株都属于同一个单一的MLVA基因型。7株玉龙菌株分离自同一疫点的不同年代,其中2006年5株,2008年1株、2009年1株。玉龙菌株与剑川的野鼠菌株有2个位点的差异,说明丽江玉龙菌株属于一个独立的基因型,与先前的生化特性比较研究的结果相对应(丽江玉龙菌株与青藏高原型菌株生化特性相近[9],与滇西纵谷型鼠疫菌关系次之,与滇闽居民型鼠疫菌关系最远[10])。
云南省的家鼠菌株属于MLVA基因Ⅱ群,大部分菌株(134株)都是一个MLVA基因型(4型),包括保山市、红河州、昆明市、临沧市、普洱市、思茅市、文山州、西双版纳州、玉溪市。这些地区分离时间跨度为1983 2002年, 这也正是发生在1982年云南省鼠疫复燃之后, 说明该基因型在云南省最近一次鼠疫流行(1982 2005年)中克隆扩张。一个特别的现象是德宏州菌株也属于4型,德宏州的菌株分离自1964 2007年间,时间前节点上早于近期云南省的鼠疫流行近期。在Ⅱ群家鼠鼠疫菌株中,被试家鼠鼠疫菌株中只有1株菌例外,该菌株(2315号)来源于红河石屏县,被分为一个MLVA基因型即5型,与其他家鼠菌株仅在M52上有1个重复数的差异,说明家鼠鼠疫菌在克隆扩张中遗传特征也在变化。
本次实验所获得的MLVA型与云南省鼠疫菌生态型以及自然疫源地的空间分布吻合较好,存在明显的地区聚集性,与菌株的分离年代没有直接的相关性。鼠疫菌的存在和型别是由其宿主、媒介和鼠疫菌相互作用的复合体,地理景观和自然环境又决定了宿主和媒介的种类及其生态学特征[11]。这也提示我们MLVA分型方法能很好的从分子水平上揭示鼠疫菌的遗传特征,数据化的表明各疫源地之间的联系,为鼠疫菌的溯源提供重要的线索,甚至可以追踪到其进化过程中的亲缘关系,在一定程度上为鼠疫的防控工作提供了有力的根据。
在VNTR的分型研究中,位点的选择对于鼠疫菌的基因分型非常重要[12],本文所筛选出的5个位点可用于云南鼠疫菌的基因分型。
参考文献
[1] Luan YQ,Yang CG,Zhang ZX,et al.Analysis on natural foci of important disease in Yunnan province from 1999 to 2008[J]. Chinese Journal of Schistosomiasis Control, 2009,21(4):311-315.(in Chinese) 栾玉泉,杨春光,张再兴,等1999——2008年云南省重要自然疫源性疾病疫情分析[J].中国血吸虫病防治杂志,2009,21(4):311-315.
[2] Song ZZ,Xia LX,Liang Y,et al.Preliminary study on plague natural foci of Yulong and ancient city in Yunnan[J]. Chinese Journal of Control of Endemic Diseases, 2008,23(1):27-31. (in Chinese) 宋志忠,夏连续,梁云,等.云南玉龙及古城区鼠疫自然疫源地判定及初步研究[J].中国地方病防治杂志,2008,23(1):27-31.
[3] Klevytska AM,Price LB,Schupp JM,et al.Identification and characterization of variable-number tandem repeats in the Yersinia pestis genome[J]. J Clin Microbiol, 2001,39(9):3179-3185.
[4] Adair DM,Worsham PL,Hill KK,et al.Diversity in a variable-number tandem repeat from Yersinia pestis [J]. J Clin Microbiol, 2000,38(4):1516-1519.
[5] Pourcel C,Andre-Mazeaud F,Neubauer H,et al.Tandem repeats analysis for the high resolution phylogenetic analysis of Yersinia pestis [J]. BMC Microbiol, 2004,4:22.
[6] Girard JM,Wagner DM,Vogler AJ,et al.Differential plague-transmission dynamics determine Yersinia pestis population genetic structure on local,regional,and global scales[J]. Proc Natl Acad Sci USA, 2004,101(22):8408-8413.
[7] Zhang XA,Hai R,Wei JC,et al.MLVA distribution characteristics of Yersinia pestis in China and the correlation analysis[J]. BMC Microbiol, 2009,9:205.
[8] Li YJ,Cui YJ,Cui BZ,et al.Features of variable number of tandem repeats in Yersinia pestis and the development of a hierarchical genotyping scheme[J]. PLoS One, 2013,8(6):e66567.
[9] Guo Y,Zhang LY,Xia LX,et al.Biochemical characteristics of Yersinia pestis in Yulong county of yunnan province[J]. Endemic Diseases Bulletin, 2008,23(3):12-14.(in Chinese) 郭英,张丽云,夏连续,等.云南省玉龙县鼠疫菌的生化特性[J].地方病通报,2008,23(3):12-14.
[10] Zhang XA.Charaeterization of Yersinia pestis isolates dispersed across China by MultiPle-LoeusVariable-NumberTandem repeat analysis[D].Beijing: Chinese Center for Disease Control and Prevention, 2008.(in Chinese) 张晓嫒.中国鼠疫耶尔森菌多位点可变数目串联重复序列分析[D].中国疾病预防控制中心,2008.
[11] Li YJ.Study of genomic polymorphism in Yersinia pestis and construction of rapid source-tracking pipeline[D].Beijing: Chinese Academy of Military Medical Sciences, 2009.(in Chinese) 李艳君.鼠疫耶尔森氏菌基因组多态性研究及快速鉴定溯源系统的建立[J].中国人民解放军军事医学科学院,2009.
[12] Fu XP,Yu DZ,Hai R.Tandem repeat sequence and its application of plague genotyping[J]. Chinese Journal of Epidemiology, 2004,25(3):269-272.(in Chinese) 付秀萍,俞东征,海荣.串联重复序列及其在鼠疫菌基因分型中的应用[J].中华流行病学杂志,2004,25(3):269-272.
[2] Song ZZ,Xia LX,Liang Y,et al.Preliminary study on plague natural foci of Yulong and ancient city in Yunnan[J]. Chinese Journal of Control of Endemic Diseases, 2008,23(1):27-31. (in Chinese) 宋志忠,夏连续,梁云,等.云南玉龙及古城区鼠疫自然疫源地判定及初步研究[J].中国地方病防治杂志,2008,23(1):27-31.
[3] Klevytska AM,Price LB,Schupp JM,et al.Identification and characterization of variable-number tandem repeats in the Yersinia pestis genome[J]. J Clin Microbiol, 2001,39(9):3179-3185.
[4] Adair DM,Worsham PL,Hill KK,et al.Diversity in a variable-number tandem repeat from Yersinia pestis [J]. J Clin Microbiol, 2000,38(4):1516-1519.
[5] Pourcel C,Andre-Mazeaud F,Neubauer H,et al.Tandem repeats analysis for the high resolution phylogenetic analysis of Yersinia pestis [J]. BMC Microbiol, 2004,4:22.
[6] Girard JM,Wagner DM,Vogler AJ,et al.Differential plague-transmission dynamics determine Yersinia pestis population genetic structure on local,regional,and global scales[J]. Proc Natl Acad Sci USA, 2004,101(22):8408-8413.
[7] Zhang XA,Hai R,Wei JC,et al.MLVA distribution characteristics of Yersinia pestis in China and the correlation analysis[J]. BMC Microbiol, 2009,9:205.
[8] Li YJ,Cui YJ,Cui BZ,et al.Features of variable number of tandem repeats in Yersinia pestis and the development of a hierarchical genotyping scheme[J]. PLoS One, 2013,8(6):e66567.
[9] Guo Y,Zhang LY,Xia LX,et al.Biochemical characteristics of Yersinia pestis in Yulong county of yunnan province[J]. Endemic Diseases Bulletin, 2008,23(3):12-14.(in Chinese) 郭英,张丽云,夏连续,等.云南省玉龙县鼠疫菌的生化特性[J].地方病通报,2008,23(3):12-14.
[10] Zhang XA.Charaeterization of Yersinia pestis isolates dispersed across China by MultiPle-LoeusVariable-NumberTandem repeat analysis[D].Beijing: Chinese Center for Disease Control and Prevention, 2008.(in Chinese) 张晓嫒.中国鼠疫耶尔森菌多位点可变数目串联重复序列分析[D].中国疾病预防控制中心,2008.
[11] Li YJ.Study of genomic polymorphism in Yersinia pestis and construction of rapid source-tracking pipeline[D].Beijing: Chinese Academy of Military Medical Sciences, 2009.(in Chinese) 李艳君.鼠疫耶尔森氏菌基因组多态性研究及快速鉴定溯源系统的建立[J].中国人民解放军军事医学科学院,2009.
[12] Fu XP,Yu DZ,Hai R.Tandem repeat sequence and its application of plague genotyping[J]. Chinese Journal of Epidemiology, 2004,25(3):269-272.(in Chinese) 付秀萍,俞东征,海荣.串联重复序列及其在鼠疫菌基因分型中的应用[J].中华流行病学杂志,2004,25(3):269-272.
|
扩展功能
|
|
| 本文信息 | |
| PDF全文 | |
| HTML全文 | |
| 参考文献 | |
| 服务与反馈 | |
| 加入引用管理器 | |
| 引用本文 | |
| Email Alert | |
| 本文作者相关文章 | |
| 朱俊洁 | |
| 王鹏 | |
| 张蓉 | |
| 海荣 | |
| 梁莹 | |
| 宋志忠 | |
| PubMed | |
| Article by ZHU Jun-jie | |
| Article by WANG Peng | |
| Article by ZHANG Rong | |
| Article by HAI Rong | |
| Article by LIANG Ying | |
| Article by SONG Zhi-zhong | |