可传播性海绵状脑病(transmissible spongiform encephalopathies,TSEs)是一类侵袭人类及多种动物中枢神经系统的退行性脑病,潜伏期长,致死率达100%。其感染因子目前认为是一种不含核酸、具有自我复制能力的感染性蛋白粒子-朊病毒(Prion),因此此类疾病又称之为朊病毒病[1]。目前已经在人类以及20余种动物中发现有自然发生或感染的TSE。人类的TSE,包括克雅氏病(Creutzfeldt-Jakob,CJD)、致死性家族型失眠症(fatal familial insomnia,FFI)、吉斯特曼-斯特劳斯综合征(gerstmann-strussler-scheinker syndrome,GSS)和库鲁病(Kurn)[2]。CJD又根据病因的不同可以分为4类:
散发型CJD(sporadic CJD, sCJD)、家族遗传型CJD(familial CJD or genetic CJD, fCJD or gCJD)、医源型CJD(iatrogenic CJD, iCJD)及变异型CJD(variant CJD, vCJD)[3]。2006年中国疾病预防控制中心(CDC )病毒病预防控制所克雅氏病监测中心开始了全国CJD(包括GSS和FFI)监测,目前监测点覆盖了12个省(直辖市、自治区)。本研究主要对我国(未包括香港、澳门和台湾地区,以下同)2012年CJD的监测数据进行总结和分析。
1 资料与方法1.1 监测体系
我国CJD的监测系统覆盖北京、上海、天津、重庆、吉林、陕西、湖北、广东、贵州、安徽、河南、新疆12个省(直辖市、自治区)。每个监测省设置1~2个哨点医院(省级三甲医院),在各省级CDC的协调下完成监测工作。哨点医院神经科医生主要负责收集临床病例资料及样品,省级CDC主要负责收集病例的流行病学资料,并将收集到的临床病例资料、流行病学资料及样品送至中国CDC病毒病预防控制所克雅氏病监测中心进行实验室检测。专家组根据病例的临床资料,脑组织、脑脊液和血液的各种检测结果进行会诊,依据《克雅氏病诊断标准》做出处理意见[4],并将最后的处理意见反馈给省级CDC,再由省级CDC反馈给相应的哨点医院。
1.2 资料来源所有CJD监测病例均按照《我国CJD监测实施方案》中的相应报告表格填写临床及流行病学信息表。其中临床病例资料主要包括病例的一般情况、首发症状、主要临床表现、临床检查(包括脑电图、睡眠运动扫描、磁共振成像)结果、样品信息及病例的死亡信息。流行病学资料主要包括居住信息、家族信息(是否在家族成员中出现过痴呆患者)、既往病史(外科手术病史及器官移植史、脑垂体提取激素使用史、输血及献血史)、特殊职业(包括医护人员、兽医及屠户)。病例诊断所收集的临床样本包括脑组织、脑脊液(CSF)及抗凝血。
1.3 样本采集及检测1.3.1 样本采集
活检及尸检的脑组织样 本,-80 ℃保存;脑脊液样本采集1~2 ml,外观透亮,无血液污染,-20 ℃保存;血液样本采集1~2 ml外周静脉的抗凝血,-20 ℃保存。所有样本需按传染性物质相关运输规定进行低温运输。
1.3.2 样本检测(1) 脑脊液中14-3-3蛋白检测 脑脊液中14-3-3蛋白阳性是CJD诊断的重要参考条件之一。取脑脊液标本20 μl,加入等量的2×SDS上样缓冲液,煮沸后按照标准操作流程进行Western blot检测,其中一抗用1 ∶ 1000稀释的14-3-3蛋白特异性多克隆抗体(Santa Cruz公司),二抗用1 ∶ 5000稀释的辣根过氧化物酶标记抗兔IgG(Thermo公司),最后用ECL检测试剂盒进行显色[5]。阳性对照采用10%羊脑组织匀浆。
(2) PRNP基因的扩增和序列分析 PRNP基因是朊蛋白基因,PRNP基因突变,可导致遗传型朊病毒病发生。人类PRNP基因129位氨基酸呈多态性分布,包括Met/Met(M/M)纯合子、Met/Val(M/V)杂合子、Val/Val(V/V)纯合子。129位氨基酸多态性与疾病易感性有关,同时参与决定疾病神经病理变化、临床病程等,129为氨基酸纯合子比杂合子更容易患sCJD。利用DNA提取试剂盒(QIAGEN公司),从外周血中提取基因组DNA。然后用PRNP基因特异性引物进行PCR扩增、测序[6],并与PRNP基因标准序列(NCBI:NM-183079.1)进行比对,检测129位氨基酸的多态性和PRNP基因是否有突变。
2 结果2.1 病例诊断分析
2012年CJD监测网络共上报CJD监测病例242例,根据《克雅氏病诊断标准》[4],发现sCJD确定诊断病例1例,脑组织中PrPSc蛋白检测为阳性;sCJD临床诊断病例63例,占报告病例的26.03%;sCJD疑似诊断病例29例,占报告病例的11.98%;fCJD或gCJD诊断病例8例,占报告病例的3.31%; 致死性家族型失眠症(D178N突变,129M/M)病例5例,占报告病例的2.07%。遗传型CJD病例突变分别为R208H、E196A和V180I各1例、E200K 2例、T188K3例。
2.2 病例流行病学特征2012年度首例病例报告日期为1月19日,最后1例病例报告日期为12月26日,病例报告无季节聚集性。
2012年总共有17个省(直辖市)有病例报送,其中病例的主要来源省份仍为监测点省份,位于前3的省份分别为北京、河南和上海。另外,非监测点省份中河北、福建、浙江、山西、江苏、甘肃、山东也有病例的上报,见图1。
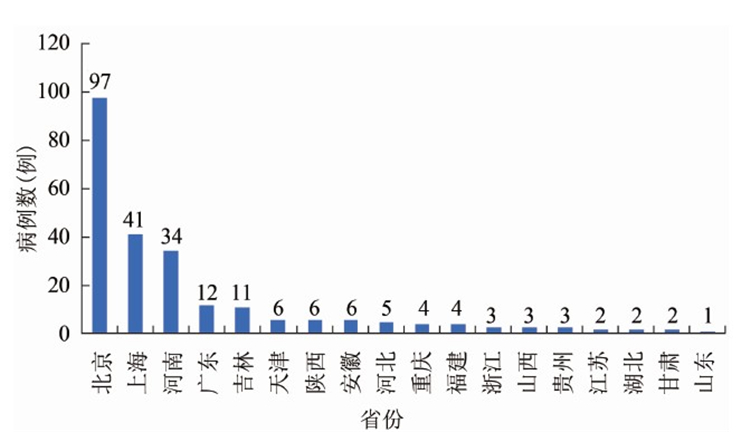
图1 2012年全国各省CJD监测病例数
Figure 1 CJD case number in China,2012
sCJD临床诊断病例年龄中值为59岁(39, 79),其中男性为30例,女性为33例,男女性别比为0.91 ∶ 1;sCJD疑似诊断病例年龄中值为53岁(36, 84),其中,男性为17例,女性为12例,男女性别比为1.41 ∶ 1。sCJD临床及疑似诊断病例的长久居住地呈散在分布,符合sCJD的发病特点。从临床及疑似病例职业分布来看,职业分布广泛,包括工人、农民、干部、教师等,见表1。
表1 sCJD临床诊断和疑似诊断病例职业分布
Table 1 Population distribution of clinical diagnosed and suspected CJD cases
| 职业 | 临床诊断例数 | 疑似诊断例数 | 合计 |
| 工人 | 12 | 4 | 16 |
| 农民 | 17 | 5 | 22 |
| 干部 | 1 | 4 | 5 |
| 教师 | 2 | 2 | 4 |
| 律师 | 0 | 1 | 1 |
| 职员 | 4 | 2 | 6 |
| 个体 | 1 | 4 | 5 |
| 退休 | 11 | 5 | 16 |
| 无业 | 15 | 2 | 17 |
sCJD临床及疑似诊断病例的首发症状以快速进行性痴呆为最多,占所有病例的46.74%,其他症状包括精神症状、小脑症状、缓慢进行性痴呆、锥体系症状、皮质性失明等,见图2。
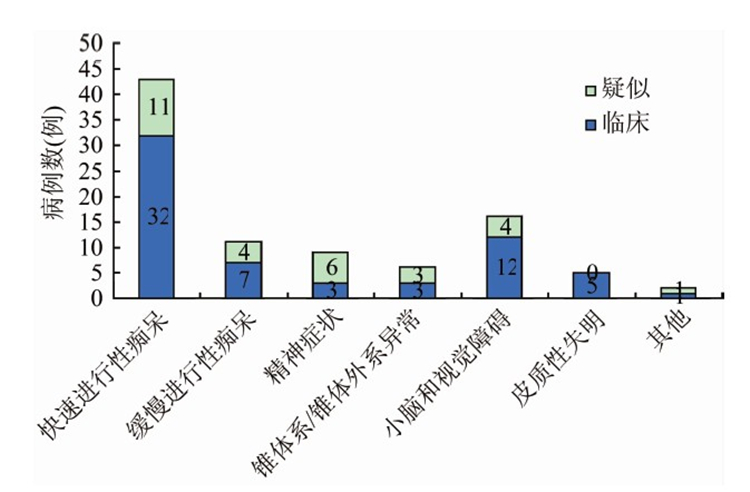
Figure 2 Initial symptoms of clinical diagnosed and suspected CJD cases
比较63例临床诊断病例和29例疑似诊断病例的临床表现,所有患者在病程的不同时期均出现进行性痴呆症状,而诊断标准中的4项典型临床表现(肌阵挛、视觉或小脑症状、锥体/锥体外系功能异 常、无动性缄默)在临床诊断病例和疑似诊断病例中出现的频率有所差异。除进行性痴呆外,在临床和疑似诊断病例中,同时出现4种临床表现,临床诊断病例为12.70%,疑似诊断病例为0;同时出现3种临床表现的在临床及疑似诊断病例中分别为46.03%和27.59%;同时出现2种临床表现的在临床及疑似诊断病例中分别为41.27%和72.41%,见表2。虽然在临床及疑似诊断病例中,大部分都仅有2种典型的临床表现,但是在2012年的监测病例中同时出现4种临床表现的病例大部分都出现在临床诊断病例中,提示CJD临床诊断病例较疑似诊断病例可能出现更多典型的临床表现。
表2 sCJD临床病例和疑似病例临床表现比较Table 2 Clinical symptoms of clinical diagnosed and suspected CJD cases
| 病例来源 | 总病 例数 |
临床表现4项 | 临床表现3项 | 临床表现2项 | |||||
| 病例数 | 构成比(%) | 病例数 | 构成比(%) | 病例数 | 构成比(%) | ||||
| 临床诊断病例 | 63 | 8 | 12.70 | 29 | 46.03 | 26 | 41.27 | ||
| 疑似诊断病例 | 29 | 0 | 0.00 | 8 | 27.59 | 21 | 72.41 | ||
脑脊液14-3-3蛋白阳性、脑电图EEG阳性以及头颅磁共振成像(MRI)典型改变均是CJD诊断的重要参考条件,我们把实验室检测结果中14-3-3蛋白阳性的病例定为A,EEG出现周期性三相波的病例定为B、头颅MRI出现特异性病变的病例定为C,见图3。3项检测中,把仅有1项检测结果阳性的定义为Ⅰ组即(A、B、C),把同时出现2项检测结果阳性的定义为Ⅱ组即(A+B、A+C、B+C),把同时出现3项检测结果阳性的定义为Ⅲ组即(A+B+C),见表3。经数据分析,在临床诊断病例中Ⅱ和Ⅲ组的病例比Ⅰ组的病例表现出更多的典型临床症状。
表3 sCJD临床诊断病例中14-3-3蛋白、EEG及MRI检测结果与临床表现的关系Table 3 Relationship between 14-3-3 protein, EE and MRI test results and clinical symptoms of clinical diagnosed CJD cases
| 分组 | 病例数 | 快速进行性痴呆 | 肌阵挛 | 视觉或小脑障碍 | 椎体/椎体外系症状 | 无动性缄默 | |||||||||
| 例数 | 百分比(%) | 例数 | 百分比(%) | 例数 | 百分比(%) | 例数 | 百分比(%) | 例数 | 百分比(%) | ||||||
| Ⅰ | 22 | 22 | 100.00 | 15 | 68.18 | 17 | 77.27 | 20 | 90.91 | 4 | 18.18 | ||||
| Ⅱ | 31 | 31 | 100.00 | 24 | 77.42 | 25 | 80.65 | 28 | 90.32 | 10 | 32.26 | ||||
| Ⅲ | 10 | 10 | 100.00 | 7 | 70.00 | 7 | 70.00 | 10 | 100.00 | 3 | 30.00 | ||||
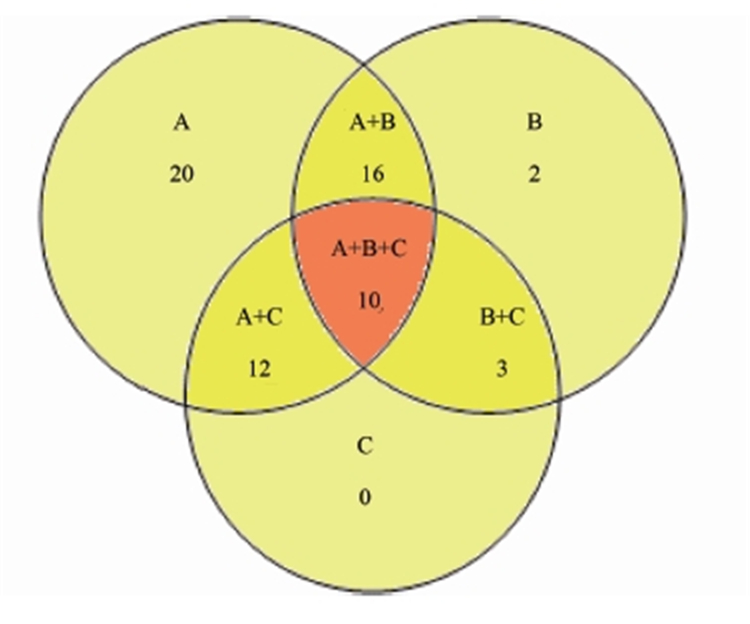
Figure 3 Number of clinical diagnosed CJD cases with positive detection results by different laboratory
在监测方案的指导下,通过2012年度全国CJD的监测工作,收集到了大量相对高质量的监测数据,完成了监测任务。根据2012年全国CJD的监测数据显示,监测病例数较2011年有所增加,其中临床诊断病例数有增加,但占总病例数的比例较2011年略有减少,整体发病情况基本符合sCJD的发病规律。
3 讨论我国CJD监测系统自2006年全面开展以来,经过几年的运行已显示了良好的监测能力。通过此监测系统,收集到包括监测点及非监测点在内的病例,并且病例的数量呈逐年增长趋势。CJD监测中心对每个病例收集到的样本(包括脑组织、脑脊液、血液)依据实验标准操作流程进行检测,并依据此检测结果经专家会诊做出最后诊断,保证了诊断结果的准确性和可靠性。
2012年通过此监测系统共上报CJD监测病例242例,其中sCJD临床诊断病例63例,sCJD疑似诊断病例29例。通过对临床及疑似病例的流行病学资料进行分析,病例的报告时间分散,没有时间的聚集性;居住地点散在,没有地区的聚集性;职业分布广泛,没有人群中的聚集性;整体符合CJD三间分布的特点。
通过对血液的PRNP检测,共发现13例具有突变的病例,占总病例的4.32%,全部突变的位点在世界上已有报道。脑脊液14-3-3蛋白和脑电图特征性改变是CJD重要的临床诊断指标[7],通常在疾病发病的早期就有相应的改变,而且同时具有两者检测阳性的病例也相对具有较多的临床症状。但是脑脊液14-3-3蛋白在发病初期可能为阴性,脑电图在发病初期也很少出现特征性的改变,这些都为诊断带来很大的困难。在CJD的发病过程中,这两个指标也会经常出现波动状态。因此,在实际工作中我们也鼓励患者在脑脊液14-3-3蛋白阴性的时候要再次进行脑脊液的检测,并且条件允许的话,进行脑电图动态监测,这对于早期发现患者有很大的意义。经临床实践,疾病早期头颅MRI呈现壳核/尾状核异常高信号,特别是弥散加权像(DW)显示对称性或不对称性皮质(或皮层)“缎带(ribbon) 征”,对于该病的早期诊断有着重要意义,目前已纳入WHO对于CJD的诊断标准中[8]。在2012年的63例临床诊断病例中,有27例在发病早期出现脑MRI检测尾状核及壳核高信号,而且其中有4例早于脑脊液14-3-3蛋白阳性的出现。通过对224份血液样品PRNP基因的检测,其中219例为M/M纯合子,占到97.77%,只有5例为M/V杂合子,此分布符合我国汉族人群129位等位基因分布特征,129位等位基因纯合子比杂合子更容易患sCJD[9][10]。
2012年监测病例中首发症状仍然是以快速进行性痴呆最多,其他的症状包括精神症状、小脑症状、缓慢进行性痴呆、锥体系症状、皮质性失明等。对于临床及疑似诊断的sCJD病例,在病程的发展过程中均会出现进行性痴呆症状,而同时具有两项症状的病例占绝大多数,具有3项症状的次之,具有4项症状的最少。临床诊断病例相对比疑似诊断病例会出现较多的典型的临床症状。综合临床诊断病例的临床症状及实验室检查结果,脑脊液14-3-3蛋白、脑电图EEG以及头颅MRI 3项检测结果中,出现阳性结果越多的病例表现出更多的典型临床表现。
目前国际上对于CJD的监测已经从发达国家逐渐扩展到发展中国家,而且国际间的交流和共享更为密切。为了提高监测质量,我国CJD监测系统也进一步加强了各监测点之间的交流和合作。遵循CJD的监测方案,进一步扩大监测网络的范围,提高监测网络的质量对于全面掌握我国CJD的发病流行状况,严密监控vCJD在我国的出现,以及最大限度地减少CJD对我国公共卫生的威胁至关重要。
[2] Liberski PP. Historical overview of prion diseases: a view from afar[J]. Folia Neuropathol, 2012, 50(1):1-12.
[3] Imran M, Mahmood S. An overview of human prion diseases[J]. Virol J, 2011, 24(8):559.
[4] WHO manual for surveillance of human transmissible spongiform encephalopathies including variant Creutzfeldt-Jakob disease. World Health Organization Communicable Disease surveillance and Response[M]. Geneva: WHO, 2003:71-72.
[5] Gao C, Han J, Zhou W, et al. Study on the characteristic of Surveillant Creutzfeldt-Jakob disease patients from January to August in 2006 in China[J]. Chinese J Exper Clin Virol, 2007, 21(3):205-207.
[6] Bratosiewicz-Wsik J, Smoleń-Dzirba J, Wataa C, et al. Association of the PRNP regulatory region polymorphisms with the occurrence of sporadic Creutzfeldt-Jakob disease[J]. Folia Neuropathol, 2012, 50(1):68-73.
[7] Matsui Y, Satoh K, Miyazaki T, et al. High sensitivity of an ELISA kit for detection of the gamma-isoform of 14-3-3 proteins: usefulness in laboratory diagnosis of human prion disease[J]. BMC Neurol, 2011, 11(1):120.
[8] Riva-AE, Jiménez-HA, Toledano R, et al. Usefulness of high b-value diffusion-weighted MRI in the diagnosis of Creutzfeldt-Jakob disease[J]. Neurologia, 2011, 26(6):331-336.
[9] Hou XS, Gao C, Zhang BY, et al. Characteristics of polymorphism of 129th amino acid in PRNP among Han and Uighur Chinese[J]. Chinese J Experi Clin Virol, 2002, 16(2):105-108.
[10] Chen C, Wang JC, Shi Q, et al. Analyses of the survival time and the influencing factors of Chinese patients with prion diseases based on the surveillance data from 2008-2011[J].PLoS One, 2013, 8(5):e62553.