Evaluation of multiple-locus variable-number tandem repeat analysis for molecular typing of Stenotrophomonas maltophilia
LI Wen-ge, TIAN Guo-zhong, LU Jin-xing
Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing 102206, China
Abstract
Objective To evaluate the applicability of multiple-locus variable-number tandem repeat analysis (MLVA) for molecular typing of Stenotrophomonas maltophilia. Methods Twelve tandem repeat loci in the total genome of S.maltophilia were analyzed with PCR and run in 2% standard agarose gel. Gels were visualized under UV light. Gel images were managed by using BioNumerics software package. Band size estimates were converted to a number of units within a character dataset with BioNumerics software. The copy numbers of twelve tandem repeat loci of 106 S.maltophilia strains was analyzed with BioNumerics (Version 4.0, Applied Maths BVBA, Belium). Results Totally 106 S. maltophilia isolates were genotyped with MLVA assay. Thirty-four MLVA genotypes were detected based on the similarity coefficient of 100%. The isolates from patients in a outbreak or from same region showed identical genotypes and could be divided into 11 groups based on the similarity coefficient of 45%. A number of MLVA clusters appeared to be partially limited to those from same geographic areas and sources. Conclusion MLVA assay is confirmed as a test which are rapid, simple, comparable, reproducible and with high discriminatory power. It can be applied in the research of molecular epidemiology of S. maltophilia.
Keywords:
Stenotrophomonas maltophilia
molecular typing
multiple-locus variable-number tandem repeat analysis
多位点串联重复序列分析方法在嗜麦芽窄食单胞菌分子分型中的应用
李文革, 田国忠, 卢金星
中国疾病预防控制中心传染病预防控制所, 北京 102206
摘要
目的 探讨多位点串联重复序列分析方法(multiple-locus variable-number tandem repeat analysis, MLVA)在嗜麦芽窄食单胞菌分型中的应用。 方法 选用文献报道的12个嗜麦芽窄食单胞菌串联重复序列位点及引物,采用聚合酶链反应(PCR)和琼脂糖凝胶电泳,根据凝胶电泳图谱计算出各位点的串联重复单元拷贝数,通过BioNumerics(Version 4.0, Applied Maths BVBA, Belium)聚类分析。 结果 设置12个位点串联重复单元拷贝数100%的相似性为判断标准,可将106株嗜麦芽窄食单胞菌分为34个MLVA基因型,流行病学上有关联的菌株具有相同的基因型。设置12个位点串联重复单元拷贝数45%相似性为判断标准,将106株菌株分为11个群,相同的地域、分离来源和分离部位的菌株具有相关性。 结论 串联重复序列位点分型技术具有简便、快速、特异、可比性、可重复性和高鉴别力等优点,适合嗜麦芽窄食单胞菌的分子流行病学研究。
内容大纲
-
1 材料与方法
- 1.1 菌株资料来源
- 1.2 试剂与仪器
- 1.3 方法
- 1.3.1 MLVA PCR扩增引物
- 1.3.2 PCR扩增
- 1.3.3 琼脂糖凝胶电泳
- 1.3.4 MLVA重复单元拷贝数和聚类分析
- 2.1 12个VNTR位点重复单元多态性
- 2.2 68株嗜麦芽窄食单胞菌MLVA分析
- 2.3 稳定性试验
多位点可变数目串联重复序列基因分型方法(multiple-locus variable-number tandem-repeat analysis,MLVA)在细菌种的水平和亚种水平上具有很好的鉴别能力,在许多病原菌的分型研究中都显示出良好的分型能力,目前国外已经建立了MLVA技术方法应用于嗜麦芽窄食单胞菌的研究报告[6]。本研究对106株嗜麦芽窄食单胞菌菌株应用MLVA技术进行分子分型的研究,以探讨该方法在嗜麦芽窄食单胞菌分型中的可应用性,旨在将该方法推广应用于临床和研究领域。
1 材料与方法
1.1 菌株资料来源106株嗜麦芽窄食单胞菌中有38株菌株引自文献[6], 68株为本实验室分离的菌株。菌株的鉴定方法包括VITEK Ⅱ 生化鉴定系统(BioMerieux, France)。
1.2 试剂与仪器
Taq DNA聚合酶,dNTP,100 bp DNA ladder Marker等PCR相关试剂购自TaKaRa公司, 仪器包括PCR扩增仪(Labcyclerk, SENAQUEST,德国), 凝胶成像仪( UVP,EC3 Darkroom,美国)。
1.3 方法
1.3.1 MLVA PCR扩增引物
MLVA PCR扩增引物序列参照文献[6],选择12个串联重复序列位点(variable-number tandem-repeat, VNTR),设计12对引物,引物序列见表1,引物由上海生物工程有限公司合成。
1.3.2 PCR扩增
25 μl PCR反应体系:10×Buffer(Mg2+,15 mmol/L) 2.5 μl,4种dNTP Mixture (均为2.5 mmol/L) 2.0 μl,10 ng DNA模板,1.0 U Taq DNA聚合酶,上、下游引物各0.8 μl(10 μmol/L),用12对引物分别进行PCR扩增。PCR反应条件:预变性95 ℃ 5 min;95 ℃ 30 s,退火温度参考表1,30 s,72 ℃ 30 s,30个循环;最后72 ℃ 5min。
1.3.3 琼脂糖凝胶电泳
PCR产物3 μl经2%的琼脂糖凝胶,电压为6 V/cm电泳2 h,经Goldview染色,100 bp DNA ladder Marker进行分子质量标记。
1.3.4 MLVA重复单元拷贝数和聚类分析
使用UVP凝胶成像分析系统中的分子量计算软件对PCR产物的DNA片段碱基含量进行测算,选择每个VNTR位点有差别两个的PCR产物进行测序,进行序列比对,换算串联重复序列的拷贝数。以12个位点的串联重复序列拷贝数为变量,采用UPGMA方法,使用BioNumerics(Version 4.0, Applied Maths BVBA, Belium) 软件进行聚类分析。每个位点的分辨力采用HGDI指标(Hunter-Gaston discrimination index,HGDI)进行计算。
2 结果
2.1 12个VNTR位点重复单元多态性 依据PCR扩增片段的大小,比较参考菌株K279a 基因组序列,测算出串联重复单元拷贝数,12个VNTR位点Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅴ、Ⅵ、Ⅶ、Ⅷ、Ⅸ、Ⅹ、Ⅺ和Ⅻ串联重复单元拷贝数见表1,106株嗜麦芽窄食单胞菌12个VNTR位点等位基因型别数分别为8、2、6、6、5、10、5、4、5、7、5和6,不同的菌株各位点的重复单元拷贝数有差别,某些菌株基因组内不存在某一串联重复单元,在Ⅵ、Ⅹ、Ⅺ和Ⅻ位点部分菌株表现为不完整的重复单元,如在Ⅵ位点,915菌株PCR扩增产物缺失了一个GTA GTG CCC GGC CGC TGG CCG GCA回文结构(SMAG结构),在嗜麦芽窄食单胞菌中该结构核苷酸序列占整个基因组核苷酸总数的0.5%;在LMG959菌株,缺失了45 bp的碱基,一个G碱基取代了SMAG回文结构,在菌株1029, 66个碱基取代了回文结构(46 bp)和10 bp碱基的核苷酸序列,导致该位点增加了10个碱基。考虑到回文结构(SMAG)是嗜麦芽窄食单胞菌DNA特征结构, 与基因重组有关,故考虑作为特殊结构,参与VNTR的聚类分析中,分别以1a、1b和1c表示,1代表该位点上存在一个串联重复单元,由于碱基的增减,导致PCR产物长度发生了变化。在Ⅻ位点,3a和3b分别表示存在3个串联重复单元,由于在该位点内核苷酸的变化,PCR扩增产物长度发生了改变。聚类分析图“XJ-sheep(44)”和“XJ-cattle(24)”中44和24表示12个位点具有相同基因型的菌株数,在Ⅺ和Ⅻ位点菌株XJ-sheep(44)和XJ-cattle(24)存在着3.5和2.5拷贝的串联重复单元,这些特殊变化,均作为变量参与聚类分析。图1为串联重复序列位点Ⅳ部分菌株凝胶电泳图谱。
2.2 68株嗜麦芽窄食单胞菌MLVA分析
以HGDI指标计算每个位点的分辨力,其中最强的为VNTR位点Ⅵ,依次为Ⅰ、Ⅹ、Ⅻ、Ⅺ、Ⅲ、Ⅳ、Ⅴ、Ⅸ、Ⅷ、Ⅶ、Ⅱ,106株 br>
表1 MLVA 12对引物序列(5′~3′)
Table 1 Primer sequences and loci of MLVA for S. maltophilia strains
| VNTR 位点 | 上游引物 下游引物 | 退火温度 (℃) | 重复单元 长度(bp) | 重复单元 拷贝数 | HGDI指标及95%可信区间 |
| Ⅰ | CAC CGC CGA GTG CGA TGC CGA TCT T ACC CGA CCG TGG ACA TGG ACG TGC G | 69 | 50 | 0,1,2,3,4,5,7,10 | 0.847(0.801~0.893) |
| Ⅱ | GAC GTG AAG TGG CTG CGC CTG AAG C CGT TCC AGC CAC TGT ACC GCC ACC A | 65 | 29 | 1,2 | 0.358(0.215~0.500) |
| Ⅲ | GTG GTG GTG ATC AAG CGC GGC AAG G GGC AGG TCG GCT GGA TGG CGG TAC T | 68 | 79 | 0,1,2,3,5,6 | 0.779(0.736~0.823) |
| Ⅳ | CAG GAA CGA TGT GCG GGC AGT GAC C CTG TCC GAA ACA CAT GGC GTG GCA G | 66 | 32 | 0,1,2,3,4,5 | 0.756(0.690~0.823) |
| Ⅴ | TGA TCG GCA TCA TCG TGG TCG GTA C GCG AGT ACC TGA GCG AAC TGG GGT G | 65 | 161 | 0,1,3,4,5 | 0.755(0.679~0.831) |
| Ⅵ | CAG CAT CAT CAA CAA GCA CCA TGG C TATC GCT TCC TGA CCA AAC CGT GGA | 65 | 106 | 0,1,2,3,4,5,6,7,8,9 | 0.917(0.875~0.958) |
| Ⅶ | GCC GCT GGT CTG GCC GTT GAT GAT G GCT GGA GCT GCA CCT GAG CGC CTG G | 65 | 37 | 0,1,2,3,4 | 0.624(0.508~0.741) |
| Ⅷ | TGG ACC GCC ACC GAC TAC CTG ATG G CAC CAC CAC CAC CGA GGT CTA CCC G | 67 | 197 | 0,1,3,4 | 0.727(0.674~0.780) |
| Ⅸ | CGA GTA CTT CAC CCC GGT CAA CGA G AGG GCC AGA CCT TCG AGG AAT TCA A | 65 | 172 | 0,1,2,4,5 | 0.746(0.662~0.830) |
| Ⅹ | TGG TGG TCA ATG ATG GGC AGC CGG A CCG CCA CGA TGA CTG GTC TCA GCC G | 66 | 67 | 0,1,3,4,5,15,17 | 0.833(0.770~0.897) |
| Ⅺ | CAC AGG TCA CAC AGC GTG GTG TAC G GGT TGT TCG GCA TCG GTT TGA TCA T | 65 | 141 | 0,1,2,3,4 | 0.815(0.754~0.876) |
| Ⅻ | CGC CAT CCA GCC GTC CTG TAC TGC T GGC GGG TTG GGT GGG TAC TAC CTG G | 66 | 103 | 0,1,2,3,4,5 | 0.824(0.763~0.886) |
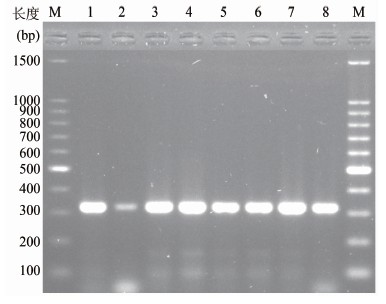
图1 8株嗜麦芽窄食单胞菌串联重复序列位点IV PCR 产物的凝胶电泳图谱
Figure 1 Agarose electrophoresis result of PCR amplification products of VNTR IV in 8 S. maltophilia isolates
嗜麦芽窄食单胞菌根据12个位点的串联重复单元拷贝数,以12个位点的拷贝数100% 相似性为判断标准,聚类分析可将106株嗜麦芽窄食单胞菌分为34个基因型;以12个位点的拷贝数45%相似性为判断标准,可将106株嗜麦芽窄食单胞菌分为11个群(C1~C11),具有流行病学关系的C1群中的OBGTC9、OBGTC10和OBGTC20菌株;C4群中916、1019和1053菌株;C5群中528和571菌株意大利分离的菌株主要在C1、C6、C7和C9群内;尼泊尔分离的菌株主要在C1、C3、C4、C5、C8和C11群内;而日本分离的菌株主要在C10群内;C7群中来自我们的68株嗜麦芽窄食单胞菌XJ-sheep(44)和XJ-cattle(24)具有相同的基因型(图2)。
2.3 稳定性试验
为了验证12个位点的稳定性,对分离的菌株 XJ-sheep(44)和XJ-cattle(24)中各选择2株菌株,连续传代培养10次,每代菌株分别提取菌株基因组DNA,对12个位点进行PCR扩增,12个位点的重复单元拷贝数没有改变。
3 讨论
嗜麦芽窄食单胞菌分型方法包括表型分型和基因分型,表型分型方法包括生物分型、血清分型、毒力试验、脂脂肪酸分型和蛋白质分析。基因分型方法主要有核糖体分型,质粒分型,16S rRNA分型,23S rRNA分型,gyrB 序列分型,限制性扩增片段长度多态性(AFLP),DNA-DNA杂交,脉冲场凝胶电泳分型(PFGE), 随机引物扩增片段长度多态性分析(RAPD), 基因间保守重复序列分型(ERIC-PCR) 和多位点序列分析(MLST)[7]。这些分型方法各有优缺点,总的说来,不是操作繁琐,就是不能数字化,MLST
图2 106株嗜麦芽窄食单胞菌MLVA聚类分析图
Figure 2 Cluster analysis of MLVA types of 106 S. maltophilia strains

虽然可以数字化,实验室内和实验室间资料具有可比性的优点,但是需要测序,提交网站,获得ST型。MLVA分子分型技术,目前日趋成熟,其优点是:操作简单,并能提供数字式的分型信息,具有很高的鉴别力和可重复性,在某种程度上与表型有对应关系。在鉴别菌株的地理来源,菌株的回溯分析和暴发事件中,具有很好的流行病学上的相关性。
本研究发现MLVA分型方法适于嗜麦芽窄食单胞菌分子流行病学研究。通过对12个VNTR位点分析,发现在某一时间,某一地区分离的菌株具有相同的基因型。分离自我国某一地区牧场牛血液中的68株菌株,具有同一基因型,说明该技术方法具有较好的分辨力和流行病学上的应用价值。意大利、尼泊尔和日本分离的菌株尽管具有不同的基因型,但可以聚集在同一群内。通过对12个VNTR位点进行2~12个位点不同的组合聚类分析发现,以12个VNTR位点聚类分析具有较好的流行病学意义。
在聚类分析上,本文应用聚类分析软件BioNumerics进行分析,该分析软件选取一定的拷贝数相似性系数,聚类结果可以分析菌株间的关联,相似性系数越大,其菌株间的流行病学关联程度越高,比较文献[6],进一步提高了其流行病学上的分析意义。本方法需要更多的菌株资料,进一步丰富菌株的分型结果和菌株的比较,可以更好的解释其流行病学意义。
参考文献
[1] Adamek M,Overhage J,Bathe S,et al. Genotyping of environmental and clinical Stenotrophomonas maltophilia isolates and their pathogenic potential[J]. PLoS One,2011,6(11):e27615.
[2] Palleroni NJ. Pseudomonas classification. A new case history in the taxonomy of gramnegative bacteria[J]. Antonie Van Leeuwenhoek,1993-1994,64(3/4):231-251.
[3] Denton M,Kerr KG. Microbiological and clinical aspects of infection associated with Stenotrophomonas maltophilia[J]. Clin Microbiol Rev,1998,11(1):57-80.
[4] Looney WJ,Narita M,Mühlemann K. Stenotrophomonas maltophilia: an emerging opportunist human pathogen[J]. Lancet Infect Dis,2009,9(5):312-323.
[5] Lipuma JJ. The changing microbial epidemiology in cystic fibrosis[J]. Clin Microbiol Rev,2010,23(2):299-323.
[6] Roscetto E,Rocco F,Carlomagno MS,et al. PCRbased rapid genotyping of Stenotrophomonas maltophilia isolates[J]. BMC Microbiol,2008,8:202.
[7] Kaiser S,Biehler K,Jonas DA. Stenotrophomonas maltophilia multilocus sequence typing scheme for inferring population structure[J]. J Bacteriol,2009,191(9):2934-2943.
[2] Palleroni NJ. Pseudomonas classification. A new case history in the taxonomy of gramnegative bacteria[J]. Antonie Van Leeuwenhoek,1993-1994,64(3/4):231-251.
[3] Denton M,Kerr KG. Microbiological and clinical aspects of infection associated with Stenotrophomonas maltophilia[J]. Clin Microbiol Rev,1998,11(1):57-80.
[4] Looney WJ,Narita M,Mühlemann K. Stenotrophomonas maltophilia: an emerging opportunist human pathogen[J]. Lancet Infect Dis,2009,9(5):312-323.
[5] Lipuma JJ. The changing microbial epidemiology in cystic fibrosis[J]. Clin Microbiol Rev,2010,23(2):299-323.
[6] Roscetto E,Rocco F,Carlomagno MS,et al. PCRbased rapid genotyping of Stenotrophomonas maltophilia isolates[J]. BMC Microbiol,2008,8:202.
[7] Kaiser S,Biehler K,Jonas DA. Stenotrophomonas maltophilia multilocus sequence typing scheme for inferring population structure[J]. J Bacteriol,2009,191(9):2934-2943.
|
扩展功能
|
|
| 本文信息 | |
| PDF全文 | |
| HTML全文 | |
| 参考文献 | |
| 服务与反馈 | |
| 加入引用管理器 | |
| 引用本文 | |
| Email Alert | |
| 本文作者相关文章 | |
| 李文革 | |
| 田国忠 | |
| 卢金星 | |
| PubMed | |
| Article by LI Wen-ge | |
| Article by TIAN Guo-zhong | |
| Article by LU Jin-xing | |