Sequence analysis of neuraminidase gene of influenza A (H1N1) pdm09 viruses in Zhejiang
CHEN Yin, MAO Hai-yan, ZHOU Min, LI Zhen, YAN Ju-ying, ZHANG Yan-jun, LU Yi-yu
Zhejiang Provincial Center for Disease Control and Prevention, Hangzhou 310051, Zhejiang, China
Abstract
Objective To understand the evolutionary characteristics of neuraminidase gene of influenza A (H1N1) pdm09 virus isolated in Zhejiang province from 2009 to early 2013. Methods Viral RNA was extracted from 29 strains of influenza A (H1N1) pdm09 virus. Neuraminidase gene was amplified by RT-PCR and the ORF sequence was assembled together by using sequencing data. Neuraminidase genes from 22 representative domestic and overseas strains during 2009-2012 were downloaded from GenBank database for analysis. All the sequences were used for the construction of phylogenetic tree by using MEGA 5.1 software. Results The 1410 bp ORF sequences of neuraminidase gene were obtained by analyzing sequencing Results of the 29 strains. After alignment, high homology was observed among all the NA genes of the strains isolated all over the world. The homology between strains isolated in Zhejiang during 2009-2012 and the representative strain of A/California/04/2009(H1N1) from North America was as high as 98.20%-99.50%. The homology between the 4 strains isolated in Zhejiang in late 2012 and early 2013 and A/California/04/2009(H1N1) strain was 98.63%-99.14%. A H275Y mutation in neuraminidase protein which could confer resistance to oseltamivir was observed in an A (H1N1) pdm09 strain isolated in January 2010 through nucleic acid sequence analysis. Conclusion Although more mutations were accumulated in NA genes in the strains isolated in Zhejiang in late epidemic stage, high homology of>98.20% was observed in all the NA genes. A H275Y mutation in neuraminidase protein which could confer resistance to oseltamivir was observed.
浙江省近年甲型H1N1流感病毒神经氨酸酶基因序列分析
陈寅, 茅海燕, 周敏, 李榛, 严菊英, 张严峻, 卢亦愚
浙江省疾病预防控制中心, 浙江 杭州 310051
摘要
目的 分析浙江省2009 2013年初甲型H1N1流感病毒神经氨酸酶基因(NA)序列进化特征。 方法 提取浙江省2009 2013年初29株甲型H1N1流感病毒基因组RNA,反转录-聚合酶链反应(RT-PCR)扩增NA基因,测序并拼接出ORF。以浙江省不同年份与地域15份代表性毒株NA序列和GenBank数据库中选取的22株2009 2012年国内外甲型H1N1流感病毒NA基因序列,采用MEGA 5.1软件对其进行序列比对并构建种系发生树。 结果 扩增并测序获得29株甲型H1N1流感病毒的NA基因ORF的全长序列,与国内外甲型H1N1流感病毒NA序列比对后显示序列的同源性较高,浙江省毒株与北美早期甲型H1N1流感病毒典型代表株A/California/04/2009(H1N1)的同源性为98.20%~99.50%,其中采集于2012年末和2013年初的4份病毒NA与A/California/04/2009(H1N1)的同源性为98.63%~99.14%。在1株采集于2010年1月的甲型H1N1流感病毒NA的基因序列中发现可导致奥司他韦耐药的H275Y突变基因型。 结论 虽然浙江省后期的甲型H1N1流感病毒神经氨酸酶基因累积了更多的变异,但所有毒株之间基因同源性仍然较高,所有毒株的神经氨酸酶基因同源性达到98.20%及以上,序列分析结果证实1份毒株携带奥司他韦耐药基因型突变。
内容大纲
-
1 材料与方法
- 1.1 材料
- 1.2 从甲型H1N1毒株中提取病毒RNA
- 1.3 RT-PCR扩增甲型H1N1病毒NA基因
- 1.4 序列测定与种系发生树的构建
- 2.1 RT-PCR扩增、测序及序列分析
- 2.2 NA基因种系发生分析
本研究选取了浙江省2009 2013年初29株甲型H1N1流感病毒流行期间的毒株,通过反转录-聚合酶链反应(reverse transcription-polymerase chain reaction,RT-PCR) 扩增并测定了NA基因序列,分析其核苷酸序列特点,为进一步开展分子生物学研究奠定基础。
1 材料与方法
1.1 材料 病毒RNA采用Qiagen RNeasy Mini Kit提取,一步法基因扩增试剂盒Titan One Tube RT-PCR System购自Roche,琼脂糖凝胶回收试剂盒购自Qiagen公司。DNA Marker DL2000和琼脂糖购自TaKaRa公司。
所选取的浙江省29株甲型H1N1流感病毒毒株分别为A/Zhejiang/2/2009(H1N1)、A/Zhejiang-Yiwu/11/2009(H1N1)、A/Haishu/SWL110/2010(H1N1),2011年初甲型H1N1流感病毒22株和2012年末和2013年初的病毒4株。其中A/Zhejiang/2/2009(H1N1)来自杭州市首例甲型H1N1流感病例,A/Zhejiang-Yiwu/11/2009(H1N1)分离自浙江省首例重症患者咽拭子样本。此外还从GenBank数据库中选取并下载了22株2009 2012年国内外毒株的NA基因,其中包括2009年早期代表性毒株A/California/04/2009(H1N1)以及来自各大洲和国内的2009 2012年毒株。
1.2 从甲型H1N1毒株中提取病毒RNA
采用Qiagen RNeasy Mini Kit提取200 μl培养细胞分离物或临床样本中的总RNA,溶于50 μl DEPC处理H2O。病毒RNA置于-70 ℃保存备用或立即进行一步法RT-PCR。
1.3 RT-PCR扩增甲型H1N1病毒NA基因
采用一对引物进行NA片段扩增,引物序列如下,NA-F: GAC CAG CAA AAG CAG GAG TT, NA-R: AGT AGA AAC AAG GAG TTT TTT GAA C,扩增产物长度1462 bp。引物由上海英潍捷基生物技术有限公司合成,用DEPC处理H2O溶解为20 μmol/L。
按Titan One Tube RT-PCR System试剂盒要求配制RT-PCR反应液,反应体系为25 μl,其中H2O 5 μl,dNTP 4 μl,DTT 2.5 μl,5× RT buffer 5 μl,2条引物各0.75 μl,Emix 1 μl,RNase Inhibitor 1 μl,病毒RNA模板5 μl。在ABI9700 PCR仪上完成扩增,扩增程序如下: 42 ℃ 30 min;94 ℃ 2 min;94 ℃ 15 s,58 ℃ 30 s,68 ℃ 90 s,循环40次; 68 ℃ 10 min。配制1.5%的琼脂糖凝胶,取扩增产物进行凝胶电泳,根据DNA分子质量标准判断扩增条带大小是否正确。选取特异性扩增条带,用琼脂糖凝胶回收试剂盒纯化目的条带。
1.4 序列测定与种系发生树的构建
纯化的PCR扩增产物由上海英潍捷基生物技术有限公司测序。除扩增引物NA-F与NA-R外,另合成两条引物用于测序,序列分别为NA-S:GAG AAC ACA AGA GTC TGA ATG TG和NA-A:GCT CCA TTA GAC GAT ACT GGA C。序列用DNAStar软件的SeqMan进行分析并拼接出NA序列。将测序的NA序列与GenBank中下载的其他地区的22条NA序列用Molecular Evolutionary Genetics Analysis version 5.1(MEGA 5.1)[3]软件进行比对,保留1410 bp的ORF区域,去除非编码区的基因序列,用Neighbor-Joining方法绘制NA基因编码区的种系发生树,采用Bootstrap法(replication=1000)进行检验。
2 结果
2.1 RT-PCR扩增、测序及序列分析 以浙江省2009 2013年初29份甲型H1N1流感病毒RNA为模板,通过RT-PCR扩增NA基因片段后电泳发现,能获得符合预期大小的特异性扩增产物。扩增产物示意图见图1。紫外灯下割取目的条带,纯化后将DNA溶于H2O中,用扩增时的2条引物和NA基因中部的2条引物同时测序。所得序列经SeqMan分析、拼接后将序列提交GenBank数据库中。基因登录号分别为A/Zhejiang/2/2009(H1N1)GQ293078、A/Zhejiang-Yiwu/11/2009(H1N1)GU108488、 A/Haishu/SWL110/2010(H1N1)HM145748。2011年初甲型H1N1流感病毒22株登录号为KC524474~KC524495,2012年末和2013年初的4株病毒登录号为KC524496~KC524499。
图1 甲型H1N1流感病毒NA基因RT-PCR扩增结果示意图
Figure 1 RT-PCR amplification results for NA gene of Influenza A (H1N1) pdm09 strains

2.2 NA基因种系发生分析
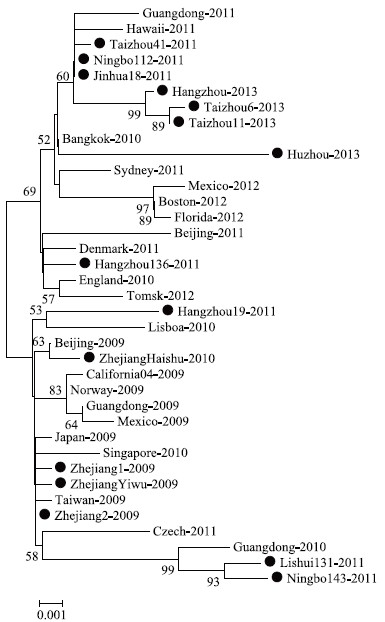
从测序的浙江省近年流感病毒NA基因中选择不同年份与地域的14条序列和1株来自浙江省首例甲型H1N1流感病例的NA基因(A/Zhejiang/1/2009(H1N1),由中国疾病预防控制中心测序),与从GenBank下载的其余22条NA序列用MEGA软件的ClustalW比对后截取共37条NA序列的ORF区域,用Neighbor-Joining方法绘制出NA基因编码区的种系发生树。序列比对结果显示,所有2009年毒株和部分2010年、2011年的毒株聚集在一个大的分支上,其余2010年、2011年的毒株和所有2012年以后的毒株位于另一分支。虽然后期的甲型H1N1流感病毒NA基因累积了更多的变异,但所有毒株之间基因同源性均较高,未发生大的变异。浙江省毒株与北美早期甲型H1N1流感病毒典型代表株A/California/04/2009(H1N1)的同源性为98.20%~99.50%,其中采集于2012年末和2013年初的4份病毒NA基因与A/ California/04/2009(H1N1)的同源性为98.63%~ 99.14%。
氨基酸水平上,浙江省的毒株NA与北美早期甲型H1N1流感病毒典型代表株A/California/04/2009(H1N1)的同源性为98.28%~99.57%之间,其中采集于2012年末和2013年初的4份病毒NA与A/California/04/2009(H1N1)的同源性除了1份为98.49%外,其余3份均为98.93%。与NA基因变异趋势类似,虽然2013年初的甲型H1N1流感病毒NA蛋白累积的氨基酸变异比平均水平略多,但仍在前几年变异水平的波动范围以内。而NA蛋白的第200位氨基酸在2012年末和2013年初的4份标本中有3个序列均发生变异,由天冬酰胺突变为异亮氨酸或丝氨酸,在其余比对的所有猪源的甲型H1N1流感病毒、人源的甲型H1N1流感病毒和此次大流行以前的人源季节性甲型H1N1流感病毒中均为天冬酰胺且非常保守。
在1株来自2010年浙江省的甲型H1N1流感毒株A/Haishu/SWL110/2010(H1N1)中发现了H275Y突变,其他浙江省毒株中未发现类似突变。NA基因种系发生树结果如图2所示。
图2 甲型H1N1病毒NA基因种系发生树
Figure 2 Phylogenetic tree of neuraminidase genes of Influenza A (H1N1) pdm09 strains
3 讨论
甲型H1N1流感疫情传播速度快,自2009年发生后甲型H1N1流感病毒迅速席卷了全球,给社会、经济和人类健康带来了严重危害。2012年末和2013年初,我国多地陆续报道甲型H1N1流感病例,虽然经过3年多的循环并已经成为季节性流感,但对甲型H1N1流感病毒监测工作依然不可或缺。
基因序列分析可以比较不同来源的流感病毒,鉴别病毒毒力和耐药性等相关的变异。此外较多的基因序列信息也大大帮助了针对流感病毒的分子检测方法的建立与评价。这些信息有助于理解流感病毒的进化和传播,并促进新型流感疫苗、新的流感诊断方法等的研究。由于NA是甲型流感病毒确定亚型的依据之一,也是通过抑制剂治疗流感的重要药物靶点,因此对甲型H1N1流感病毒的NA基因序列进行分析具有重要意义。此外由于NA的编码基因有较高的变异性,对其变异与进化分析尤为重要。
从同源性分析结果分析,与早期毒株相比,虽然2010 2012年的甲型H1N1流感病毒NA基因累积了更多的变异,但所有毒株之间基因同源性均较高,未发生大的变异。在1株来自2010年初浙江省的甲型H1N1流感毒株A/Haishu/SWL110/2010(H1N1)中发现了H275Y突变,其他浙江省毒株中未发现类似突变。出现变异的毒株分离自1名29岁的女性患者,发病前一直居于当地,无外出史。经过对同时期同地域病毒的检测未发现其他类似变异毒株。我国于2009年曾经在1名由国外入境的感染甲型H1N1流感病毒的人员中鉴定出H275Y突变基因型。
目前,已经确认的甲型H1N1流感病毒突变导致的耐药性变化大多为H275Y变异[4],但随着疫情的发展、研究的加强和对样本序列信息的分析的深入,NA基因中其他位点的突变在国外也渐有报道。对于2009年甲型H1N1大流行之前的季节性H1N1流感病毒,由于变异导致在2008年出现了奥司他韦耐药病毒比例的急剧提升 ,甚至某些地区2008 2009年初的流感季节中季节性H1N1流感病毒的耐药比例达到100% 。2009年至今甲型H1N1病毒绝大部分仍然对奥司他韦敏感,但由于流感病毒的变异频率和传播能力较强,因此对于甲型H1N1流感病毒NA基因的序列分析并进行变异监测不可或缺。此外,在基因型监测的基础上同时也不可忽视对于耐药表型的监测,通过酶学活性检测可以直接了解病毒的耐药能力,同时也可以实现对非常见位点变异的监测。由于甲型H1N1流感病毒对于M2离子通道抑制剂(金刚烷胺类药物)普遍耐药,对于NA基因分子层面的分析还将有助于防控策略的制定和指导患者治疗,因此通过NA基因序列的分析从而了解病毒对NA抑制剂的敏感性显得尤为重要。
本研究通过对浙江省甲型H1N1流感病毒的NA进行基因序列和进化分析,进一步了解病毒的变异及其分子特征,对从基因水平上深入认识甲型H1N1病毒,监测病毒的突变与变异提供了依据。
参考文献
[1] Guo YJ, Cheng XW. Influenza virus and its experimental technology[M]. Beijing:China Three Gorges Publishing House, 1997.(in Chinese) 郭元吉,程小雯.流行性感冒病毒及实验技术[M].北京:中国三峡出版社,1997.
[2] Huang L, Zhou JF, Wei H, et al. Neuraminidase inhibitors resistance in influenza viruses and the related mechanisms[J].Chinese Journal of Virology,2012,28(5):572-576.(in Chinese) 黄兰,周剑芳,韦红,等.流感病毒神经氨酸酶抑制剂药物耐药现状及机制研究进展[J].病毒学报,2012,28(5):572-576.
[3] Tamura K, Peterson D, Peterson N, et al. MEGA5:molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods[J].Mol Biol Evol,2011,28(10):2731-2739.
[4] van der Vries E, Jonges M, Herfst S, et al. Evaluation of a rapid molecular algorithm for detection of pandemic influenza A(H1N1) 2009 virus and screening for a key oseltamivir resistance(H275Y) substitution in neuraminidase[J].J Clin Virol,2009,47(1):34-37.
[5] Besselaar TG, Naidoo D, Buys A, et al. Widespread oseltamivir resistance in influenza A viruses(H1N1), South Africa[J].Emerg Infect Dis,2008,14:1809-1810.
[6] Sheu TG, Deyde VM, Okomo-Adhiambo M, et al. Surveillance for neuraminidase inhibitor resistance among human influenza A and B viruses circulating worldwide from 2004 to 2008[J].Antimicrob. Agents Chemother,2008,52(9):3284-3292.
[7] Hurt AC, Ernest J, Deng YM, et al. Emergence and spread of oseltamivir-resistant A(H1N1) influenza viruses in Oceania, South East Asia and South Africa[J].Antiviral Res,2009,83(1):90-93.
[8] Moscona A. Global transmission of oseltamivir-resistant influenza[J].N Engl J Med,2009,360(10):953-956.
[2] Huang L, Zhou JF, Wei H, et al. Neuraminidase inhibitors resistance in influenza viruses and the related mechanisms[J].Chinese Journal of Virology,2012,28(5):572-576.(in Chinese) 黄兰,周剑芳,韦红,等.流感病毒神经氨酸酶抑制剂药物耐药现状及机制研究进展[J].病毒学报,2012,28(5):572-576.
[3] Tamura K, Peterson D, Peterson N, et al. MEGA5:molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods[J].Mol Biol Evol,2011,28(10):2731-2739.
[4] van der Vries E, Jonges M, Herfst S, et al. Evaluation of a rapid molecular algorithm for detection of pandemic influenza A(H1N1) 2009 virus and screening for a key oseltamivir resistance(H275Y) substitution in neuraminidase[J].J Clin Virol,2009,47(1):34-37.
[5] Besselaar TG, Naidoo D, Buys A, et al. Widespread oseltamivir resistance in influenza A viruses(H1N1), South Africa[J].Emerg Infect Dis,2008,14:1809-1810.
[6] Sheu TG, Deyde VM, Okomo-Adhiambo M, et al. Surveillance for neuraminidase inhibitor resistance among human influenza A and B viruses circulating worldwide from 2004 to 2008[J].Antimicrob. Agents Chemother,2008,52(9):3284-3292.
[7] Hurt AC, Ernest J, Deng YM, et al. Emergence and spread of oseltamivir-resistant A(H1N1) influenza viruses in Oceania, South East Asia and South Africa[J].Antiviral Res,2009,83(1):90-93.
[8] Moscona A. Global transmission of oseltamivir-resistant influenza[J].N Engl J Med,2009,360(10):953-956.
|
扩展功能
|
|
| 本文信息 | |
| PDF全文 | |
| HTML全文 | |
| 参考文献 | |
| 服务与反馈 | |
| 加入引用管理器 | |
| 引用本文 | |
| Email Alert | |
| 本文作者相关文章 | |
| 陈寅 | |
| 茅海燕 | |
| 周敏 | |
| 李榛 | |
| 严菊英 | |
| 张严峻 | |
| 卢亦愚 | |
| PubMed | |
| Article by CHEN Yin | |
| Article by MAO Hai-yan | |
| Article by ZHOU Min | |
| Article by LI Zhen | |
| Article by YAN Ju-ying | |
| Article by ZHANG Yan-jun | |
| Article by LU Yi-yu | |